
Effect of Methanol Extract of A. Bonnei on Some Neuroprotective Parameters/Markers (Vitamin E, Adenine Deaminase and Acetylcholinesterase) in Wistar Rats
- Aroh Samuel Chinekwu
- Winnifred Njideka Adiri
- Promise Udoka Asogwa
- Bruno Basil
- Ireh Princes Chioma
- Okereke Chiamaka Edith
- 1105-1127
- Aug 21, 2024
- Microbiology
Effect of Methanol Extract of A. Bonnei on Some Neuroprotective Parameters/Markers (Vitamin E, Adenine Deaminase and Acetylcholinesterase) in Wistar Rats
Aroh Samuel Chinekwu, Winnifred Njideka Adiri, Promise Udoka Asogwa, Bruno Basil, Ireh Princes Chioma, Okereke Chiamaka Edith
University of Nigeria Nsukka, Nigeria
DOI: https://doi.org/10.51244/IJRSI.2024.1107088
Received: 02 July 2024; Revised: 13 July 2024; Accepted: 17 July 2024; Published: 21 August 2024
ABSTRACT
The aim of this work is to evaluate the effect of methanol extract of A. bonnei on some neuroprotective parameters/markers (vitamin E, adenine deaminase and acetylcholinesterase) in Wistar Rats. The leaves of the plant A. bonnei were collected at Diogbe, Igbo-Etiti local government area of Abia state. They were air dried, milled and stored in an air tight container for further use. A total of 16 adult Wistar rats of male sex (100–130 g) used for this study were acclimatized for one week and the rats were randomly divided into four equal groups. Group 1 (normal control) received only water and feed. Group 2 served as the positive control (and received mercury chloride). Group 3 was intoxicated with mercury chloride and treated with Diazepam (5 mg/kg). Animals in group 4 were neurointoxicated and treated with the plant extract (400 mg/k). From the results of vitamin E, in the cerebellum, it was observed group 3 when compared to the group 1, showed a significant (p<0.05) increase; while group 2 on comparism to group 1, showed a significant (p<0.05) decrease. Also, group 4 showed an insignificant (p >0.05) on comparism with group 1. Furthermore, the group 2 on comparism to the group 3, indicated a significant decrease (p<0.05); while group 4, showed an insignificant decrease when compared with group 3. In the cerebrum, From the chart above, the untreated group when compared to the normal control, showed a significant (p<0.05) decrease; while the intoxicant + A. bonnie group when compared to the normal control, had an insignificant (p>0.05) decrease. Furthermore, when the untreated group was compared to the intoxicant + standard drug group, a significant (p<0.05) decrease was recorded; while the intoxicant + A. bonnie group in comparism with the intoxicant + standard drug group, showed an insignificant (p<0.05) decrease. From fig. 4.2 showing result of acetylcholinesterase, in the cerebellum, the untreated group in comparism to the normal control, had a significant (p<0.05) increase, while the intoxicant + extract group when compared with the normal control had an insignificant (p<0.05) decrease. Also, the untreated group on comparism to the intoxicant + standard drug group, recorded a significant (p<0.05) increase; while the intoxicant + extract group when compared to the intoxicant + standard drug group had an insignificant (p>0.05) increase. Considering the cerebrum, the untreated group when compared to the normal control, showed a significant (p<0.05) increase, while there was a significant decrease when intoxicant + extract group was compared to the normal control. Furthermore, the untreated group when compared to the intoxicant + standard drug group showed a significant (p<0.05) increase; while there was a significant decrease when the intoxicant + extract group was compared to the intoxicant + standard drug group. Finally, the result of adenine deaminase showed that in the cerebellum, there was an insignificant (p>0.05) decrease existed when the untreated group was compared with the normal control; also the intoxicant + extract group showed an insignificant (p>0.05) decrease on comparism to the normal control. Furthermore, an insignificant (p>0.05) decrease was shown when the untreated group was compared to the intoxicant + standard drug group; while an insignificant (p<0.05) increase was recorded when the intoxicant + extract group was compared to the intoxicant + standard drug. In the cerebrum, a significant (p>0.05) decrease was shown when the untreated group was compared with the normal control; while a significant (p<0.05) increase was recorded when the intoxicant + extract group was compared to the normal control. Also, an insignificant (p>0.05) decrease was recorded when the untreated was compared to the intoxicant + standard drug group; while a significant increase (p<0.05) was shown for intoxicant + extract group against the intoxicant + standard drug group. In this work, it has been seen that mercury intoxication affects the cerebrum and cerebellum of animals. From these results, it has been observed that the methanol leaves extract of A. bonnei, possesses some neuroprotective properties.
INTRODUCTION
The brain can be described as the most complex structure in the human body. It is made up of neurons and neuroglia, the neurons being responsible for sending and receiving nerve impulses or signals. The microglia and astrocytes are essential for ensuring proper functioning of neurons. They are quick to intervene when neurons become injured or stressed. As they are sentinels of neuron well-being, pathological impairment of microglia or astrocytes could have devastating consequences for brain function. It is assumed that neuroglial activation is largely determined by neuronal signals (Halliwell, 1992). Acute injury causes neurons to generate signals that inform neuroglia about the neuronal status. Depending on how severe a degree of neuronal injury, neuroglia will either nurse the injured neurons into regeneration or kill them if they are not viable. These types of neuroglial responses are considered to represent normal physiological and neuroprotective responses. In contrast, some processes that are chronic in nature persistently activate neuroglia eventually causing a failure in their physiological ability to maintain homeostasis. This could have detrimental consequences and may lead to bystander damage due to neuroglial dysfunction.
Neurodegeneration is a process involved in both neuropathological conditions and brain ageing. It is known that brain pathology in the form of cerebrovascular and neurodegenerative disease is a leading cause of death all over the world, with an incidence of about 2/1000 and an 8% total death rate (Kolominsky et al, 1998). Cognitive dysfunction, is a major health problem in the 21st century, and many neuropsychiatric disorders and neurodegenerative disorders, such as schizophrenia, depression, Alzheimer’s Disease (AD) dementia, cerebrovascular impairment, seizure disorders, head injury, Parkinsonism etc can be severely functionally debilitating in nature (Commenges et al., 2000). Neuroprotection refers to the strategies and relative mechanisms able to defend the central nervous system (CNS) against neuronal injury due to both acute (e.g. stroke or trauma) and chronic neurodegenerative disorders (e.g. Alzheimer’s disease and Parkinson’s disease) (Kumar, 2006). Moreover, stroke and dementia are a source of high individual and family suffering mainly because of the lack of efficient therapeutic alternatives. The latter motivates research efforts to identify the mechanisms of neuronal death and to discover new compounds to control them.
Herbal medicine has long been used to treat neural symptoms. Although the precise mechanisms of action of herbal drugs have yet to be determined, some of them have been shown to exert anti-infl ammatory and/or antioxidant effects in a variety of peripheral systems. Now, as increasing evidence indicates that neuroglia-derived chronic inflammatory responses play a pathological role in the central nervous system, anti-inflammatory herbal medicine and its constituents are being proved to be a potent neuroprotector against various brain pathologies. Structural diversity of medicinal herbs makes them a valuable source of novel lead compounds against therapeutic targets that are newly discovered by genomics, proteomics, and high-throughput screening.
Medicinal plant is defined as any plant with one or more of its organs containing substance that can be used for therapeutic purpose or which can be used as precursors for the synthesis of antimicrobial drugs (Bouayed et al., 2007). Plants are presently the sources of medicines for many people of different age in many country of the world, where diseases are treated primarily with traditional medicines obtained from plants. The modern pharmaceutical industry itself still relies largely on the diversity of secondary metabolites in plants and secondary metabolites of which at least 12,000 have been isolated; a number estimated to be less than 10% of the total (Mallikharjuna et al., 2007). In plants, the synthesized aromatic substances (metabolites) are used as defensive weapons against predation by microorganisms, insects and herbivores.
Alstonia boonei, a large evergreen tree belonging to the family Apocynaceae is one of the widely used medicinal plants in Africa and beyond. The important plants of genus Alstonia includes Alstonia scholaris, Alstonia boonei, Alstonia congensis and Alstonia macrophylla which have proved to be useful in various diseases. Almost all plant parts viz. leaves, stem bark; root and inflorescences have been used and are further under investigative study. It is distributed throughout the tropics and the rain forest of west and Central Africa (Olever-Bever, 1986; Olajide et al., 2000). It is known by different names in different cultures and tribal settings. It is not edible as food but possess roots, stems, barks, leaves fruits, seeds, flowers, and latex which are claimed to have medicinal properties in some cultures.
Statement of the Problem
Phytopharmaceuticals are gaining importance as modern medicine as well as traditional system of medicine owing to their therapeutic potential. Novel antioxidants may offer an effective and safe means of bolstering body’s defense against free radicals (Rajadurai and Prince, 2006) and thereby provide protection against AD like problems. Various natural antioxidants like curcumin, rosmarinic acid, huperzine A have been reported to have a neuroprotective effect against AD (Frank and Gupta, 2005; Kumar et al., 2007; Alkam et al., 2007). Alstonia boonei stem bark has been reported to have an antipsychotic effect in murine model. Other parts of the plant has been shown to have other beneficial properties such as antioxidant, sedative, anti-snake venom, etc. Therefore, this present study was designed to investigate the possible neuroprotective effect of Alstonia boonei against mercury chloride-induced cognitive impairment and associated oxidative damage in rats.
Aim of the Study
The aim of this study is to investigate the neuroprotective properties of ethanol leaf extract of Alstonia boonei in wistar rats.
Objectives of the Study
The objectives of this study is to evaluate the effect of the plant extract on the following parameters: vitamin E, acetylcholinesterase and adenine deaminase in Wistar rats.
LITERATURE REVIEW
Traditional Plants
Many cultures throughout the world still rely on indigenous medicinal plants for their primary health care needs (Halliwell, 1992). To date, 25% of modern medicines are derived from plants that have been used by traditional medical practitioners (Kolominsky et al., 1998). It is a fact that traditional systems of medicine have become a topic of global importance. Although modern medicine may be available in many developed countries, people are still turning to alternative or complementary therapies including medicinal herbs. Yet, few plant species that provide medicinal herbs have been scientifically evaluated for their possible medical applications. The safety and efficacy data are available for even fewer herbs, their extracts and active ingredients and the preparation containing them. Tropical and subtropical Africa contains between 40–45,000 species of plant with a potential for development and out of which 5,000 species are used medicinally (Commenges et al., 2000). Still there is a paradox, in spite of this huge potential and diversity, the African continent has only contributed 83 of the 1100 classic drugs globally (Commenges et al., 2000). African countries are at a stage where traditional medicine is considered more for its capacity to generate other medicine than for its own sake. In many cases research undertakings and the commercial use stemming from that research have always relied on information provided by the local communities and, in many instances, have hardly benefited from the research results (Kumar et al., 2006). In Africa, traditional healers and remedies made from plants play an impokrtant role in the health of millions of people. The relative ratios of traditional practitioners and university-trained doctors in relation to the whole population in African countries are revealing. In Ghana, for example, in the Kwahu district, there are 224 people for every traditional practitioner, compared to nearly 21,000 people for one university-trained doctor (Kumar et al., 2006).
Alstonia boonei

Figure 1: Plate showing clockwise order from top right, the leaves, stem, branches, and whole plant of Alstonia boonei John et al., 2012
Typically, studies on the medicinal plants such as Alstonia boonei, Abbiw, (1990) have focused on the bioactivity of its chemical constituents, ethnobotany, pharmacology, and taxonomy. However, a comprehensive or systematic review on the plant is lacking. Furthermore, in much of the older literature concerning West Africa, the name Alstonia congensis has been erroneously used for Alstonia boonei. Consequently, this paper synthesizes studies on Alstonia boonei. This is necessary to recapitulate the findings on the plant and thereby provide a comprehensive and current repository for references on the plant.
Alstonia comprises about 40 species and has a pantropical distribution. There are about twelve species of the genus Alstonia. Alstonia boonei De Wild belongs to the family Apocynaceae. The species are scattered all over the world of which two are indigenous to Africa. The plant is known locally in Ghana as Onyame dua, Osen-nuru, or Sinduro in Twi, Onyame dua in Fante, Sinu or Adawura in Ga-Adangbe, Bakunin, Nyamenlebaka, Emenle, or Emie in Nzema, and Siaketekre, Nyemi dua, or Asi atoe in Ewe (Ayiku, 1992). Elsewhere, Alstonia is known as Australian fever bush, Australian quinine, Devil tree, Dita bark, fever bark, or palimara (Gosse et al., 1996). It grows into a giant tree in most of the evergreen rain forests of tropical West Africa. The plant thrives very well in damp riverbanks. It is well known to all the traditional healers practicing along the west coast of Africa. It occurs in deciduous and fringing forest of Ghana (Gosse et al., 1996). Alstonia boonei De Wild is a deciduous tree up to 35 meters high. It buttresses deep-fluted high and narrow. Its white latexes are copious. The leaves are in whorls at nodes, oblanceolate, apex rounded to acuminate, lateral vein prominent almost at right angle to midrib. The flowers are white with lax terminal cymes. The fruits are paired with slender follicle up to 16 cm long with brown floss at each end.
Ethnobotanical Uses
The bark of Alstonia tree is one of the effective analgesic (Abbiw et al., 1990) herbs available in nature. All the parts of the plant are very useful but the thick bark cut from the matured tree is the part that is most commonly used for therapeutic purposes.The bark of the tree is highly effective when it is used in its fresh form; however, the dried one could equally be used. Therapeutically, the bark has been found to possess antirheumatic (Abbiw et al., 1990), anti-inflammatory (Abbiw et al., 1990), analgesic/pain-killing, antimalaria/antipyretic, antidiabetic (mild hypoglycaemic), antihelminthic, antimicrobial and antibiotic properties (Hadi and Bremner, 2001). A decoction could be sweetened with pure honey and be taken up to 4 times daily as an effective painkiller for the following conditions.
Painful menstruation (dysmenorrhoea), when associated with uterine fibroid or ovarian cysts in women; lower abdominal and pelvic congestion associated with gynaecological problems such as pelvic inflammatory diseases; to relieve the painful urethritis common with gonococcus or other microbial infections in men. Alstonia decoction also exerts a mild antibacterial effect in this case, relieving the aches and pains associated with malaria fever. Alstonia is taken in the form of preparations that exhibits antipyrexia and anti-malaria effects, to combat rheumatic and arthritic pains. The decoction of Alstonia bark could be taken alone as an effective pain-killing agent. A cold infusion made from the fresh or dried bark of Alstonia taken orally two to three times daily exerts a mild hypoglycaemic effect on diabetic patients. The cold infusion is also administered orally for the purpose of expelling round worms, threadworms (Abbiw et al., 1990), and other intestinal parasites in children.
The fresh bark of Alstonia could be used in preparing herbal tinctures; it is particularly useful as an effective antidote against snake, rat, or scorpion poison. It is also useful in expelling retained products of conception and afterbirth when given to women. Asthma can be treated with a drink prepared from parts of Trema orientalis and decoction of the bark of Alstonia boonei mixed with the roots and bark of cola and fruits of Xylopia parviflora with hard potash (Abbiw et al., 1990). The bark decoction of Alstonia boonei is used with other preparations in the treatment of fractures or dislocation (Abbiw et al., 1990), jaundice, and for inducing breast milk. Its latex is taken as a purgative. The hardened latex is used for the treatment of yaws. Alstonia boonei De Wild is regarded as one of few herbs with potential anti-HIV indicators. In some African countries Alstonia boonei is considered a sacred tree and worshiped in the forest and hence human beings in those countries do not eat its parts.
The flowers of Alstonia scholaris contain n-hexacosane, lupeol, β-amyrin, palmitic acid, and ursolic acid. The components were extracted with petroleum ether at 60–80°C and separated by chromatographic methods. Mass spectra and spectrographic methods were used for identification. The root and root bark of Alstonia scholaris contain α-amyrin, α-amyrin acetate, lupeol acetate, stigmasterol, β-sitosterol, and campesterol and its isomer Desoky et al., 2000). The stem bark of Alstonia scholaris contains α-amyrin acetate, lupeol acetate, and β-sitosterol (Khaleque et al., 1991; Kweifio-Okai, 1993). The isolation of α-amyrin acetate and lupeol acetate was done with 96% ethanol on the air-dried bark. The concentrated extract was stored for 2 weeks and gave a solid, which (on several crystallizations from EtOH) gave colorless needles of melting point of 160°C. This product was chromatographed through a column of alumina (4 × 32 cm) and eluted with petroleum ether (boiling point 40–60°C). Rechromatography of the fractions gave 2 products. The first one forms colourless plates in (EtOH-Et2O) with mpt of 224–5°C, [α]29D 80° (all rotations detected in CHCl3), with one acetyl group and no active H. The second product colourless needles in (EtOH-Et2O) with mpt of 215–16°C, [α]28D 40°, containing an acetyl group and no active H. On hydrolysis of the first product with methanolic KOH, a substance with mpt of 184°C, [α]28D 84°, identical with α-amyrin was obtained. This on benzoylation gave α-amyrin benzoate which forms prisms in (C6H6-EtOH) with mpt of 198°C, [α]28D 92°. Similarly, on hydrolysis the second product gave a substance with mpt 212–13°C, [α]27D 22.6°, identical with lupeol. Benzoylation gave lupeol benzoate which forms colourless plates in (C6H6-EtOH) with mpt of 259–60°C, [α]27D 60.4°.
Pharmacological Properties
Triterpene compounds (R1 = H, C ≥ 10 fatty acid acyl) are useful as anti-inflammatory and antiarthritic agents (Kweifio-Okai and Carroll, 1992). α-Amyrin acetate isolated from petroleum ether extract of Alstonia boonei root bark was hydrolyzed with NaOH to α-amyrin followed by esterification with palmitoyl chloride to obtain α-amyrin palmitate. Rats were injected with 150 μL of complete Freund’s adjuvant containing 10mg/mL Mycobacterium tuberculosis in the right hind footpad. From days 11–19 rats were fed orally with 56 mg I/kg in 1 mL of water. Regression analysis of the rate of the ankle diameter change from days 11–19, postadjuvant injection showed that the diameter of the ankle decreased by 31% in treated animals.
In a similar research work, the triterpenes-α-amyrin acetate, β-amyrin acetate, β-amyrin, and lupeol acetate isolated from the petroleum ether extract of Alstonia boonei De Wild, root barks were tested for their anti-inflammatory effects in CFA-induced arthritic rats (Kweifio-Okai and Carroll, 1993). When administered orally daily from days 11 to 19 after adjuvant, lupeol acetate and β-amyrin acetate were most effective in preventing further increases in ankle adjuvant swelling. All triterpenes abrogated the increases in WBC count, increased liver and/or kidney weights but only α-amyrin acetate significantly increased serum GOT levels. In the presence of β-amyrin, there was significant neutrophil degeneration. Triterpenes of Astonia boonei root barks were shown to be antiarthritic but the effects on liver and kidney weights raised the possibility of toxicity in antiarthritic therapy.
In another development, lupeol acetate isolated from the petroleum ether fraction of Alstonia boonei root barks was tested for its anti-arthritic effect in CFA-induced arthritic rats (Rajic et al., 2000). It was administered orally every 48 hrs (66 mg/kg body wt.) from days 32 to 40 after adjuvant and assessed on day 60. Lupeol acetate was able to return the increase in spleen weight and the reduction in serum alkyl phosphatase to nonarthritic control values.
The anti-inflammatory triterpenoids are also inhibitors of serine proteases (Olajide et al., 2000). The lupane triterpenoid lupeol, the ursane triterpenoid alpha-amyrin, and esters of these compounds present in the bark of roots of Alstonia boonei (Apocynaceae) have anti-inflammatory properties. Alpha-Amyrin is a competitive inhibitor of bovine trypsin and chymotrypsin (Ki values 29 microM and 18 microM, resp.). Lupeol linoleate, lupeol palmitate, and alpha-amyrin linoleate are noncompetitive inhibitors of trypsin (Ki values 7 microM, 10 microM, and 16 microM, resp.). Alpha-amyrin linoleate is also a non-competitive inhibitor of chymotrypsin (Ki value 28 microM). Lupeol is a competitive inhibitor of both trypsin and chymotrypsin (Ki values 22 and 8 microM, resp.). Alpha-amyrin palmitate is a potent non-competitive inhibitor of chymotrypsin (Ki 6 microM). Lupeol, alpha-amyrin, and the palmitic and linoleic acid esters of these compounds are ineffective or very weak as inhibitors of porcine pancreatic elastase and of Lucilia cuprina and Helicoverpa punctigera leucine aminopeptidases. These hydrophobic triterpenoids represent further examples of anti-inflammatory triterpenoids that are PKA inhibitors as well as being selective protease inhibitors.
When the methanol extract of the stem bark of Alstonia boonei was investigated for anti-inflammatory, analgesic, and antipyretic properties (Kweifio-Okai, 1991), it was found out that the extract caused a significant (P < 0.05) inhibition of the carrageenan-induced paw oedema, cotton pellet granuloma, and exhibited an anti-arthritic activity in rats. Vascular permeability induced by acetic acid in the peritoneum of mice was also inhibited. The extract also produced marked analgesic activity by reduction of writhing induced by acetic acid, as well as the early and late phases of paw licking in mice. A significant (P < 0.05) reduction in hyperpyrexia in mice was also produced by the extract. This study has established anti-inflammatory, analgesic, and antipyretic activities of the stem bark of Alstonia boonei.
Mercury
More than 2500 A.C., the prehistoric man used the cinabrio (mercury sulfide), due to its red-gold color, to draw on cave walls and perform face painting. Subsequently, mercury has been used in the amalgamation (direct burning of metallic mercury on the gravel, promoting the separation of gold), in photography and as an antiseptic in the treatment of syphilis (Ayiku 1992; Gosse et al., 1999). Exposure to mercury brought harmful effects to health of humans, but changes resulting from human exposure to mercury only called the attention of the scientific society after the accidents in Japan and Iraq (Abbiw, 1990). In Japan, a serious accident occurred resulting from the deposition of industrial waste with large quantities of mercury in the Minamata Bay. Mercury was then ingested by human through fish intake, thus triggering signs and symptoms such as ataxia, speech impairment, visual field constriction, sensory disturbance, deafness, blindness, tremors, involuntary movements, mental retardation, coma, and death. Infants whose mothers were infected developed mental retardation, peripheral neuropathy, cerebral palsy, and blindness.
Forms of Mercury Exposure
Mercury is now considered an environmental pollutant of high risk to public health because of its high toxicity and mobility in ecosystems ( Kucera et al., 1972). Exposure to mercury can occur from both natural and artificial sources. Human activities that can result in mercury exposure include the burning of fossil fuels, chlor-alkali industries, mining, the burning of waste, and the use of coal and petroleum (Rahman et al., 1985; Kam et al., 1997).
More natural sources of mercury include volcanic activity, earthquakes, erosion, and the volatilization of mercury present in the marine environment and vegetation (Kam et al., 1997; Alexandre, 2006; and Lindsberg et al., 2007). Mercury emitted both naturally or as a result of human activity is primarily found as inorganic metal vapor (Hg0) (Boening, 2000). Among the natural sources of mercury, the largest emissions are from the degassing of the earth’s crust. More than five tons of mercury is estimated to be released into the sea every year as a result of erosion and geochemical cycles (Swain et al., 2007).
Mercury contaminates the environment through a cycle involving the initial emission, the subsequent atmospheric circulation of the vapor form, and the eventual return of mercury to the land and water via precipitation (Alexandre, 2006). The emission of mercury is an important part of this cycle of contamination and can occur through natural processes or as a result of human activities, as mentioned above (Lindsberg et al., 2007).
Mercury present in seas and rivers after methylation can contaminate fish (EPA, 1997; Hansen et al., 1997). The consumption of fish contaminated with mercury is a major source of mercury exposure in the Amazon basin. Studies show that the concentration of mercury in the muscles of fish that are widely consumed in the Amazon region are greater than the limit set by WHO (World Health Organization) as safe for human consumption (0.5 g/kg) (Kam et al., 1997).
Effect of Mercury on the Central Nervous System(CNS)
Among the compounds of mercury, the methylmercury is primarily responsible for the neurological alterations present in humans and experimental animals. It is believed that the mechanisms are related to the toxic increase in reactive oxygen species (ROS). Oxidative stress is associated with the etiology of neurodegenerative diseases such as amyotrophic lateral sclerosis, Parkinson’s disease, and Alzheimer’s disease (Roulet et al., 1998; and Bridges et al., 2010), but these mechanisms have yet to be fully recognized. Reinforcing the hypothesis that the majority of injuries caused by methylmercury (MeHg) in the central nervous system are related to its ability to increase reactive oxygen species, Zhang et al. (2009) reported that after pretreatment of bovine cells with pyrroloquinoline quinone (PQQ), an antioxidant, the cytotoxicity induced by MeHg is significantly attenuated. PQQ reduces the percentage of apoptotic cells, decreased significantly ROS production, suppressed lipid peroxidation, and increased antioxidant enzyme activity in cells exposed to MeHg. Furthermore, the protective effects elicited by an antioxidant (ebselen) strengthen the idea that seleno-organic compounds represent promising approaches to neutralize MeHg-induced neurotoxicity (Yin et al., 2011).
Studies also demonstrate that mercury has the ability to reduce the number of neuron and cytoarchitecture in individuals with prenatal exposure to mercury (Grandjean et al., 1997; Graeme et al., 1998). In animal models, some of these symptoms are reproduced. Low-dose prenatal exposure to methylmercury during 10 gestational days impairs motor and mnemonic function in adult mice (Kweapradub et al., 1997). This hypothesis is supported by studies that describe methylmercury inhibition of cell division and migration both “in vivo” and “in vitro” (Grandjean et al., 1997; Grandjean et al., 1999). In addition, because of its high affinity for sulfhydryl groups in tubulin, methylmercury inhibits the organization of microtubules that are important in CNS development (Ponce et al., 1994; Kishimoto et al., 1995). The binding to SH groups also interferes with the intracellular signaling ofmultiple receptors (e.g.,muscarinic, nicotinic, and dopaminergic) and promotes the blockade of Ca++ channels in neurons (Castoldi et al. 2000). In addition, inorganic mercury has the ability to increase the permeability of chloride channels of GABA A receptors in the dorsal root ganglion, which is associated with neuronal hyperpolarization (Mottet et al., 1997).
Corroborating these findings, the study conducted by Maia et al., (2010) demonstrates that the poisoning by methylmercury changes the nitrergic activities of adult mice, and the predominance of alterations may be related to different locations. Besides increasing the nitrergic activity methylmercury and mercuric chloride also have the ability to increase the release of neurotransmitters such as acetylcholine, dopamine, norepinephrine, and serotonin.
Similar findings have also been reported to be a mechanism implicated in the effects of methylmercury and HgCl2 on the central nervous system function (Weinsberg et al., 1995; Haung et al. 1996). Halbach et al. studied a correlation in Iraqi children between the level of maternal exposure to methylmercury during pregnancy and psychomotor retardation. Sandborgh-Englund et al. corroborated this finding in children from the Faroe Islands; they found that children exposed to mercury in the prenatal period had defects in attention, memory, language, and motor function. In addition, exposure to methylmercury in pregnant women or early childhood leads to changes in the CNS development of the fetus or child, respectively (Geier et al., 2003). Thereupon, changes caused by mercury poisoning result in significant clinical deficit in motor skills, coordination, and general activity rate of cognitive and psychological disorders (Kaewpradub et al., 1997).
Effect of Mercury on the Cardiovascular System
For decades, the toxic effects of mercury were associated mainly with the central nervous system; however, inorganic mercury also produces profound cardiotoxicity (Hussain et al., 1997; Brawer et al., 1998). Halbach and collaborators showed that mercury concentrations in hair reached up to 150 μg/g in populations living in the Amazon basin. Furthermore, nearly all of the inhabitants of 40 cities studied have blood concentrations above the reference values. In this population, it has been demonstrated that exposure to mercury by frequent consumption of fish has a strong positive correlation with increased arterial blood pressure (Halbach, 1990). Other studies also correlate mercury exposure with increased risk of hypertension, myocardial infarction, coronary dysfunction, and atherosclerosis (Bastos et al., 2006; Fillions et al., 2006). Data presented by Yoshizawa et al. showed that mercury exposure was associated with the progression of atherosclerosis and an increased risk of developing cardiovascular disease. Houston followed patients for approximately 13.9 years and found an association between the concentration of mercury in the hair and the risk of developing cardiovascular events or dying from cardiovascular disease and other causes.
The mechanism by which mercury produces toxic effects on the cardiovascular system is not fully elucidated, but this mechanism is believed to involve an increase in oxidative stress. Exposure to mercury increases the production of free radicals, potentially because of the role of mercury in the Fenton reaction (Ehara et al., 2001; Mackness et al., 2004) and a reduction in the activity of antioxidant enzymes, such as glutathione peroxidase. The MeHg reaction with the glutathione peroxidase occurs via thiol (–SH) and/or selenol (–SeH) groups from endogenous molecules (Farina et al., 2011). Even though there are 4 of glutathione molecules containing selene in their active sites, only the cytoplasmic glutathione peroxidase 1 (GPx 1) changes hydrogen peroxide to water (Arthur, 2000). The reduction in glutathione peroxidase with selenium-dependent activity is the result of the decreased bioavailability of selenium, a molecule that is required for enzymatic activity (Magos et al., 2006). The high affinity of mercury to the thiol group can lead to decreased glutathione peroxidase selenium-dependent activity. Other antioxidant enzymes which participate against reactive oxygen species due to mercury intoxication are catalase and superoxide dismutase.
The increment of ROS and reduction of the antioxidant activity increase the risk of developing cardiovascular disease (Ganther, 1980; Valko et al., 2006). Sherwani et al. (2011) showed that MeHg has the capacity to induce phospholipase D (PLD) activation through oxidative stress and thiol-redox alterations. They investigated the mechanism of the MeHg-induced PLD activation through the upstream regulation by phospholipase A2 (PLA2) and lipid oxygenases such as cyclooxygenase (COX) and lipoxygenase (LOX) in the bovine pulmonary artery endothelial cells. Their results showed that MeHg significantly activates both PLA2 and PLD. MeHg also induces the formation of COX- and LOX-catalyzed eicosanoids in endothelial cells.
Cardiovascular changes resulting from mercury poisoning are also described in animal models. However, the mechanism involved in the effects of mercury on the cardiovascular system is not fully understood but seems to be dependent on both the dose and time of exposure. Raymond and Ralston studied the hemodynamic effects of an intravenous injection of HgCl2 (5 mg/kg) in rats and observed that mercury produced cardiac diastolic failure and pulmonary hypertension. Moreover, Naganuma et al. [ reported that acute exposure to HgCl2 (680 ng/kg) increased blood pressure, heart rate, and vascular reactivity to phenylephrine in rats; this increased reactivity seems to depend on an increased generation of free radicals. Perfused hearts from animals exposed acutely to HgCl2 showed a reduction in left ventricular systolic pressure, heart rate, and atrioventricular conduction delay (Rossoni et al., 199).
Acute Brain Injury
Acute brain injury, whatever its cause, is associated with considerable short-term and long-term morbidity and mortality. In the USA it is estimated that 52,000 fatalities arise as a result of traumatic brain injury (TBI) every year, and approximately 5.3 million people live with TBI-related disabilities (Selassie et al., 2008). These figures are similar in the European Union, where an estimated 7.7 million people have TBI-related disabilities (Tagliaferri et al., 2006). Accurate data from emerging economies, where TBI is increasing due to greater motorization, are lacking, but are likely to be similar or worse (Roozenbeek et al., 2013). Stroke is the second leading cause of death and the third leading cause of disability-adjusted life-years worldwide, and its global burden is increasing (Hankey, 2013). Other ischemic and hemorrhagic lesions to the brain, such as subarachnoid hemorrhage or ischemia–reperfusion after cardiac arrest, are also associated with high mortality and devastating sequelae (Zacharia et al., 2010; Koenig, 2014).
Following the primary cerebral insult, a cascade of events amplifies the initial damage regardless of the etiology of the precipitating event. Secondary biochemical changes contribute to subsequent tissue damage with associated neuronal cell death. The time course over which these effects occur may be longer than assumed previously, potentially providing a wider time window for interventions. Neuroprotective agents that can limit secondary tissue loss and/or improve behavioral outcomes have been identified in multiple animal models of acute brain injury. However, translation to the clinical setting has been largely disappointing.
Therapeutic modalities for neuroprotection
1. Reperfusion strategies in ischemic stroke
Early reperfusion therapy to restore blood flow to salvageable ischemic brain can prevent cell death and facilitate neurological recovery. Reperfusion may be accomplished through administration of intravenous thrombolytic agents, intra-arterial thrombolysis, mechanical thromboembolectomy, ultrasound-enhanced thrombolysis and various combinations of these approaches. Alternative or adjunct approaches include enhanced oxygen delivery, hemodilution and systemic central hemodynamic augmentation therapy (Barreto and Alexandrov, 2012). Risks associated with reperfusion therapies, apart from failure to reperfuse, include intracerebral hemorrhage (ICH), ischemia–reperfusion injury and catheterization complications. A large-scale randomized trial indicated that intravenous recombinant tissue plasminogen activator (alteplase) administered within 3 hours of ischemic stroke onset was associated with significantly improved functional outcomes when compared with placebo, but with an increased risk of ICH (NINDS, 1995). More recent data indicated that intravenous alteplase was beneficial when given within 4.5 hours of onset in nondiabetic patients <80 years old without massive strokes (Hacke et al., 2008).
Despite increased risks of ICH and early death, a Cochrane review of 27 controlled trials of thrombolytic agents given within 6 hours of acute ischemic stroke onset indicated improved survival and neurological outcome at 3 to 6 months; benefits were greater in patients treated within 3 hours (Wardlaw et al., 2014). The evidence offered by these papers has been incorporated into guidelines, which recommend early intravenous thrombolysis (Jauch et al., 2013).
2. Autoregulation and neuroprotection
Cerebrovascular autoregulation is the ability of the brain to maintain a constant cerebral blood flow (CBF) through a range of cerebral perfusion pressures (CPP) (Rangel-Castilla et al., 2008). Several dynamic pressure reactivity indices have been proposed for monitoring cerebrovascular autoregulation in real time at the bedside, by calculating the correlation between the arterial blood pressure and continuous measures of CBF or cerebral blood volume (Rangel-Castilla et al., 2008). When this correlation is negative or close to zero, autoregulation is assumed to be present (pressure active); when it is positive, autoregulation is considered to be absent (pressure passive). ICP, transcranial Doppler, brain tissue oxygenation and near-infrared spectroscopy have all been used as estimates of global CBF/cerebral blood volume. Accordingly, the CPP range at which autoregulation is best preserved (optimal CPP) can be estimated. However, whether a strategy based on these measurements might be used to improve outcomes has yet to be demonstrated (Steiner et al., 2002).
3. Hemoglobin management for neuroprotection
Anemia is common among patients with severe brain injury and is associated with poor outcomes in TBI, aneurysmal subarachnoid hemorrhage (SAH), ICH and acute ischemic stroke (Kramer and Zygun, 2009; LeRouxm 2013). In the absence of serious cardiac disease, a restrictive red blood cell transfusion strategy (for example, trigger hemoglobin (Hb) 7 g/dl) is usually recommended in critically ill patients (Retter et al., 2013). In severe brain injury, however, compromised brain tissue oxygenation may occur at higher Hb levels than in other ICU patients (Hebert et al., 1999; Le Roux, 2011).
In a recent randomized clinical trial in 200 patients with closed head injury, erythropoietin was compared with placebo in patients with transfusion thresholds of 7 g/dl versus 10 g/dl (Robertson et al., 2014). There were no statistically significant differences in 6-month neurological outcomes between the two transfusion groups, but the higher transfusion threshold was associated with more adverse events. Based on these limited numbers of patients, it seems premature to conclude that a low transfusion threshold may be beneficial in all TBI patients; more data are needed.
Novel therapeutic modalities
Neurorepair strategies
In addition to neurological damage, acute brain injury induces a series of neurorestorative events (Lo, 2008). In some cases, the central nervous system is able to remodel itself following insults that impair tissue homeostasis. Neurorestorative events include neurogenesis, gliogenesis, angiogenesis, synaptic plasticity and axonal sprouting. These processes are stimulated by endogenous growth-related factors and may continue for weeks to months, facilitating functional and structural recovery. Unfortunately, these restorative processes are largely ineffective for the severity of damage usually encountered in TBI or stroke. Accordingly, providing such injured tissue with a milieu that enhances neuroregenerative processes has become an important therapeutic target.
Mesenchymal stromal cells
Infusion of mesenchymal stromal cells (MSCs) can improve structural and functional outcomes in different brain injury models (Laroni et al., 2013). MSCs secrete growth and neurotrophic factors or induce their production by resident brain cells, including microglia. The interaction between MSCs and inflammatory microenvironments is crucial. MSCs can reprogram the local microenvironment from a detrimental function to a beneficial role, reducing toxic events and promoting endogenous restorative processes (Phinney and Sensebe, 2013; Zanier et al., 2014).
Remote ischemic conditioning
Brief repeated cycles of peripheral vascular occlusion and de-occlusion in dogs prior to induction of coronary ischemia reduce myocardial infarct size (Murry, 1986). This remote ischemic preconditioning is presumed to induce humoral factors that prevent reperfusion injury in several organs, including the brain. Protection occurs through modification of intracellular kinase activity, mitochondrial permeability and the inflammatory response to reperfusion (Kharbanda et al., 2009). One potential means by which ischemic conditioning can be achieved is by application of a standard blood pressure cuff to the arm and alternating 5-minute cycles of inflation and release (Kharbanda et al., 2002)
Ischemic conditioning may be applied before (preconditioning), during (perconditioning) or after (postconditioning) a cerebral ischemic event. In patients, perconditioning as an adjunct to treatment with intravenous alteplase was associated with a reduction in tissue risk of infarction after acute thrombotic stroke (Hougaard et al., 2014). Preconditioning has also been associated with prevention of recurrent stroke in patients with intracranial arterial stenosis (Meng et al., 2012). Nevertheless, several questions remain unanswered: would remote ischemic conditioning reduce infarct size if administered before thrombolytic reperfusion of acute thrombotic stroke; can repeated (daily for weeks to months) ischemic conditioning improve long-term outcomes after cerebral ischemia; and in what other settings could ischemic conditioning reduce reperfusion injury and produce better clinical outcomes?
Volatile anesthetic agents for neuroprotection
Volatile anesthetic agents may have neuroprotective properties. Pretreatment with isoflurane improved long-term neurological outcomes after experimental hypoxic/ischemic bran injury or focal brain ischemia (McAuliffe et al., 2007) and post-treatment provided neuroprotection in rats (Segal et al., 2012). The inducible form of nitric oxide synthase may mediate the tolerance to ischemia. Other factors that could be involved are the inhibition of excitatory neurotransmission and regulation of intracellular calcium responses during ischemia. Although attractive for their potential benefit on ischemic damage, these experiments have not been translated into clinical studies because use of a volatile agent may also induce vasodilatation, increasing CBF and, consequently, elevations in ICP.
In SAH patients, however, in whom an increase in CBF may be beneficial, the availability of a pragmatic bedside dispensing device and continuous ICP and regional CBF monitoring has made the inhalation of isoflurane for neuroprotection in the ICU practical, and this approach has been assessed in a small pilot study (Villa et al., 2012). Nevertheless, at this stage, the use of volatile agents remains unproven and not ready for clinical implementation (Bosel et al., 2012).
Hyperoxia in neuroprotection
Oxygen is an essential substrate for the brain; however, the safety margin between effective and toxic oxygen doses is relatively narrow. Oxygen may be toxic to the lungs (for example, tracheobronchitis, absorption atelectasis, hypoxic pulmonary vasoconstriction and hyperoxia-induced lung injury), the circulation and the brain tissue itself (seizures or lipid peroxidation) (Lambertsen et al., 1953), and hyperoxia has been associated with increased mortality in patients with various acute neurological disease processes (Kilgannon et al., 2010; Brenner et al., 2012; Rincon et al., 2014). However, hyperoxia can increase PbtO2, restore mitochondrial redox potential, decrease ICP, restore aerobic metabolism and improve pressure autoregulation (Tolias et al., 2004; Nortje et al., 2008; Rockswold et al., 2010). Administering 100% oxygen at normal atmospheric pressure (normobaric hyperoxia) is inexpensive, widely available and can be started promptly after TBI or stroke (for example, by paramedics). Results in humans, however, have been mixed (Tolias et al., 2004; Diringer et al., 2007; Quintard et al., 2014). There is probably a narrow effective dose, and benefit may be limited to at-risk tissue. Moreover, treatment may only be effective in specific subgroups of patients or may depend on the metabolic state (Vilalta et ql., 2011). Furthermore, whether improvements in brain metabolism translate into better outcome is unclear (Quintard et al., 2014).
Hyperbaric oxygen has been shown to reduce infarct volume, blood–brain barrier disruption, edema and neurologic deficits in animal models of ischemic brain injury (Miljkovic-Lolic et al., 2003). In experimental TBI, hyperbaric oxygen decreased neuron injury and edema (Vlodavsky et al., 2006). In a small group of severe TBI patients, hyperbaric oxygen improved brain metabolism and decreased ICP (Rockswold et al., 2001). However, administering hyperbaric oxygen can be clinically challenging, requiring that patients are moved out of the ICU to the hyperbaric oxygen chamber, an expensive facility available in only a few centers. In a small study of 42 TBI patients, the combination of hyperbaric and normobaric hyperoxia was associated with significant outcome benefits compared with standard therapy (Rockswold et al., 2013).
MATERIALS AND METHOD
Materials
A. Equipment and Apparatus
- Blender
- Filter paper
- funnel
- Water bath
- Beakers
- Stirrer blender (manual)
- Measuring cylinder (pyrex, england)
- spatula
- Centrifuge
- Sample bottles (plain)
- Pipette (Pyrex, England)
- Electronic weighing balance (sartorius sensitive weighing balance (1200g)
- Digital weighing balance
- Gavage
- Syringes
- Test tubes
B. Chemicals
- Mercury chloride
- Methanol (99%) volume
- Diazepam (5mg)
- Bioun solution
C. Plant Materials
The leaves of the plant Anthocleista vogelii were collected from the matured plant at Umudike, Ikwuano local government area of Abia state in the month of June 2018. The plant sample was authenticated at the herbarium, Department of Plant Science and Biotechnology (PSB), Michael Okpara University of Agriculture Umudike (MOUAU), Abia state, Nigeria. And a herbarum sample was deposited.
Extraction of Plant Materials
The plant extract as gotten following a stepwise procedure, first about 500g of A. Vogelii was collected, weighed and air dried. This was followed by oven drying and then grinding the leaves. After grinding, 100g of it was weighed out on a beaker and soaked in 700mls of methanol for 72 hours, after which it was extracted and filtered into the beaker using a filter paper. The filtrate was then concentrated using a water bath at temperature of 40-50oC until the methanol was fully evaporated leaving only the gel like green substance which is the plant extract. It was properly labeled and stored in the refrigerator at 4°C until used.
Experimental Animals
Sixteen (16) adult Wistar albino rats of male sex (100–130 g) used in the study were acquired from the animal unit of the College of Veterinary Medicine, Michael Okpara University of Agriculture, Umudike, Abia State, Nigeria. The animals were fed with standard feed (Vital feeds finisher), provided with free access to water under a well-ventilated condition of 12hrs light cycle. They were kept in aluminum cages and were allowed to acclimatize for two weeks before the commencement of the experiments. The study was carried out in accordance with the Organization for Economic and Development (OECD) principles on Good Laboratory Practice (GLP) (OECD 2001). Prior ethical approval (Code number, UMSE/06/012) was obtained from the ethical committee on the use of animals of the College of Veterinary Medicine, Michael Opkara University of Agriculture, Umudike, Nigeria.
Experimental Design
The rats were randomly divided into four groups consisting of four rats each. Group 1 (normal control) consisted of normal rats that neither received mercury chloride nor any drug (water and feed only). Group 2 served as the positive control (neurotoxicant control). Rats in group 3 were induced with neurotoxicant (mercury (ii) chloride) and treated with Diazepam at normal dose (5mg/kg). Animals in group 4 were neurointoxicated and treated with the plant extract at high dosage (400 mg/kg).
Standard drugs and sample used
The standard drugs and aqueous extract of Anthocleista vogelii were prepared by directly weighing and dissolving the known weight of the drugs and solid extract (after evaporation of methanol) in the required volume of di-ionized water and tween 80. The standard drug (Diazepam) was obtained from a pharmaceutical shop in Umuahia, Abia State.
Induction of neurotoxicant
A cohort of male Wistar rats was fed for at least 8 hours. Neurotoxicant was induced in each rat by administering Mercury chloride (4mg/ Kg body weight; orally) in distilled water. The control cohort was administered normal water orally. At 7days post-induction of neurotoxicant, neurotoxicity was assayed by observation method on hair loss signs and swollenness of animal body parts particularly the lower jaw.
Treatment of neurodegenerative rats with oral neurotoxicant drug
Oral neurotoxicant drug (Diazepam) was used in the present study. The drug was administered orally to a cohort of neurointoxicated rats (n=4) at 7:00 – 9:00 each day for 28 days. Diazepam was administered at 5mg/kg body weight/day. Untreated neurointoxicated group received only the vehicle (distilled water).
Assay of Biochemical Parameters
Assay of Serum Acetylcholinesterase
The activity of AChE in the brain was determined by the method described by Ellman et al., (1961) as modified by Srikumar et al., (2004). The mixture was prepared by mixing 0.4 ml aliquot of the homogenate and added to 2.6 ml phosphate buffer (0.1 M, pH 8.0) and 100 μL of DTNB (270 μM). This was pre-incubated for 2 minute at 30oC and the reaction was started with the addition of 20 μL ATC (30 mM). The product of thiocholine reaction with DTNB was determined at 412nm for a period of 10 minute at 2 minute intervals. The absorbance per minute was determined. The specific activity is expressed as μmol ACTC min-1 mg protein-1.
Assay of Serum Vitamin E
Preparation of standard curve
Working standard of tocopherol (27g/mL): 1 mL of stock standard solution was taken and made the volume up to 100 mL with ethanol (aldehyde free) to obtain a concentration of 27g/mL. This solution is stable at room temperature. Six centrifuge tubes were taken and labeled as B (blank) and S 1 , S 2 , S 3 , S 4 , and S 5 for standard solution. In these tubes, 0 (zero), 150,300, 450, 600, and 750L of working standard solution (27 g/mL) were added, respectively, and made the volume up to 750L by adding ethanol (aldehyde free). These solutions (S 1 – S 5 ) are equivalent to 4, 8, 12, 14, 16, and 20g/mL of tocopherol concentration, respectively. The absorbance was measured by using 200L of each of the above-mentioned solutions (including blank) put on a plain ELISA microplate (non-antibody coated) and read in an ELISA reader (ERBA-Lisa Scan II) at 492nm.
Adenine deaminase
Pipet 0.5 ml. of buffered substrate, pH 7.03, into each of 2 test tubes (15 X 125-mm.). Mark one tube “sample” and the other “blank.” Place both tubes in a 370 water bath for 2-3 min. Add to the sample tube 0.05 ml. of unhenmolyzed serum or plasma. Stopper, mix gently, and replace in water bath. Exactly 60 min. after adding the serum, add 2.5 ml. of phemiol color reagent to each tube. This stops the reaction. To the blank tube, add 0.05 ml. of serum and to both tubes add 2.3 ml. of alkali-hypochlorite reagent. Stopper amid promptly mix by inverting at least 3 times. Place both tubes in a heating block or water bath for 15 mm. for color development. Readings are made in 10mm. round cuvets in a spectrophotometer, at 640 mn., against the reagent blank adjusted to 100% transmittance. The color is stable for about 20 mins. The units of adenosine deaminase activity are read from the calibration curve. One unit of adenosine deaminase activity is the amount that will liberate 1 /Lg. of ammonia nitrogen per milliliter of serum/plasma per hour at 37o. To convert these units to international units, multiply by 0.476 (Giusti et al., 1984).
RESULTS AND INTERPRETATION
Effect of plant extract of A. bonnei on vitamin E in the cerebellum and cerebrum of Wistar albino rats
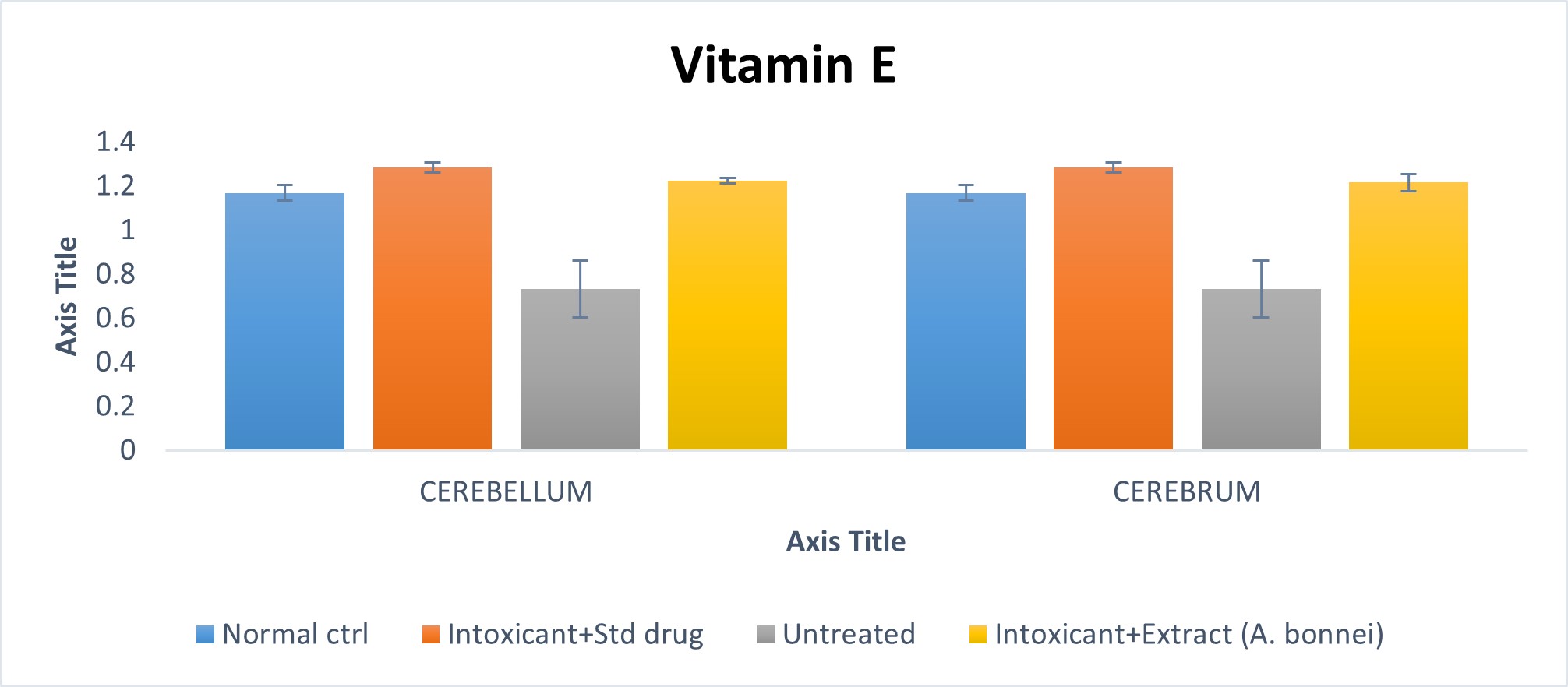
Fig. 4.1 Effect of plant extract of A. bonnei on vitamin E in the cerebellum and cerebrum of Wistar albino rats.
Cerebellum
Using the chart above, the intoxicant+ standard drug group when compared to the normal control, showed a significant (p<0.05) increase; while the untreated group on comparism to the normal control, was shown to have a significant (p<0.05) decrease. Also, the intoxicant + A. bonnie treated group showed an insignificant (p >0.05) on comparism to the normal control. Furthermore, the untreated group on comparism to the intoxicant + standard drug group, indicated a significant decrease (p<0.05); while the intoxicant + A. bonnie group, showed an insignificant decrease when compared with the intoxicant + standard drug group.
Cerebrum
From the chart above, the untreated group when compared to the normal control, showed a significant (p<0.05) decrease; while the intoxicant + A. bonnie group when compared to the normal control, had an insignificant (p>0.05) decrease. Furthermore, when the untreated group was compared to the intoxicant + standard drug group, a significant (p<0.05) decrease was recorded; while the intoxicant + A. bonnie group in comparism with the intoxicant + standard drug group, showed an insignificant (p<0.05) decrease.
Effect of plant extract of A. bonnei on Acetylcholine esterase in the cerebellum and cerebrum of Wistar albino rats
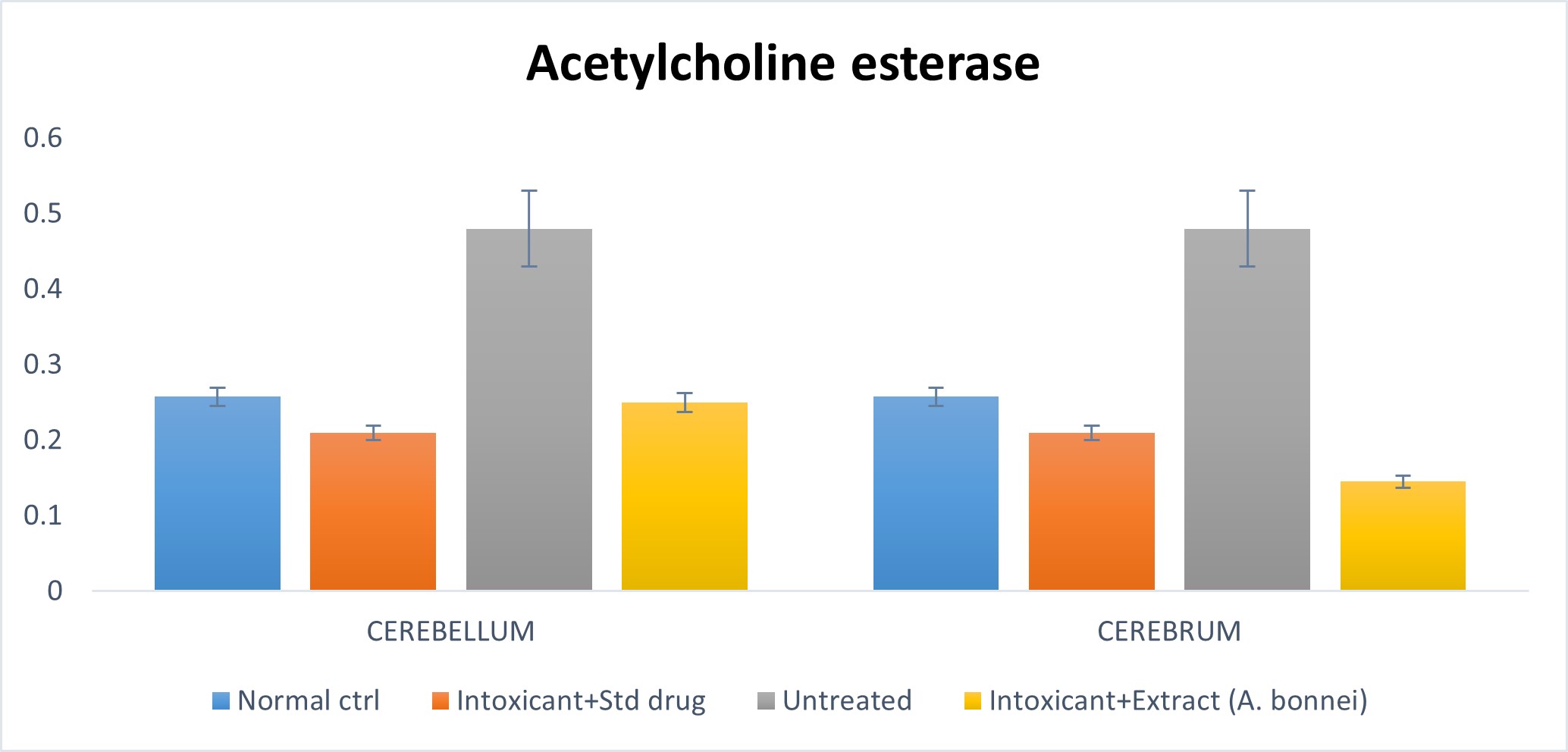
Fig. 4.2: Effect of plant extract of A. bonnei on Acetylcholine esterase in the cerebellum and cerebrum of Wistar albino rats
Cerebellum
Using the chart above, the untreated group in comparism to the normal control, had a significant (p<0.05) increase, while the intoxicant + extract group when compared with the normal control had an insignificant (p<0.05) decrease. Also, the untreated group on comparism to the intoxicant + standard drug group, recorded a significant (p<0.05) increase; while the intoxicant + extract group when compared to the intoxicant + standard drug group had an insignificant () increase.
Cerebrum
From the chart above, the untreated group when compared to the normal control, showed a significant (p<0.05) increase, while there was a significant decrease when intoxicant + extract group was compared to the normal control. Furthermore, the untreated group when compared to the intoxicant + standard drug group showed a significant (p<0.05) increase; while there was a significant decrease when the intoxicant + extract group was compared to the intoxicant + standard drug group.
Effect of plant extract of A. bonnei on Adenine deaminase in the cerebellum and cerebrum of Wistar albino rats
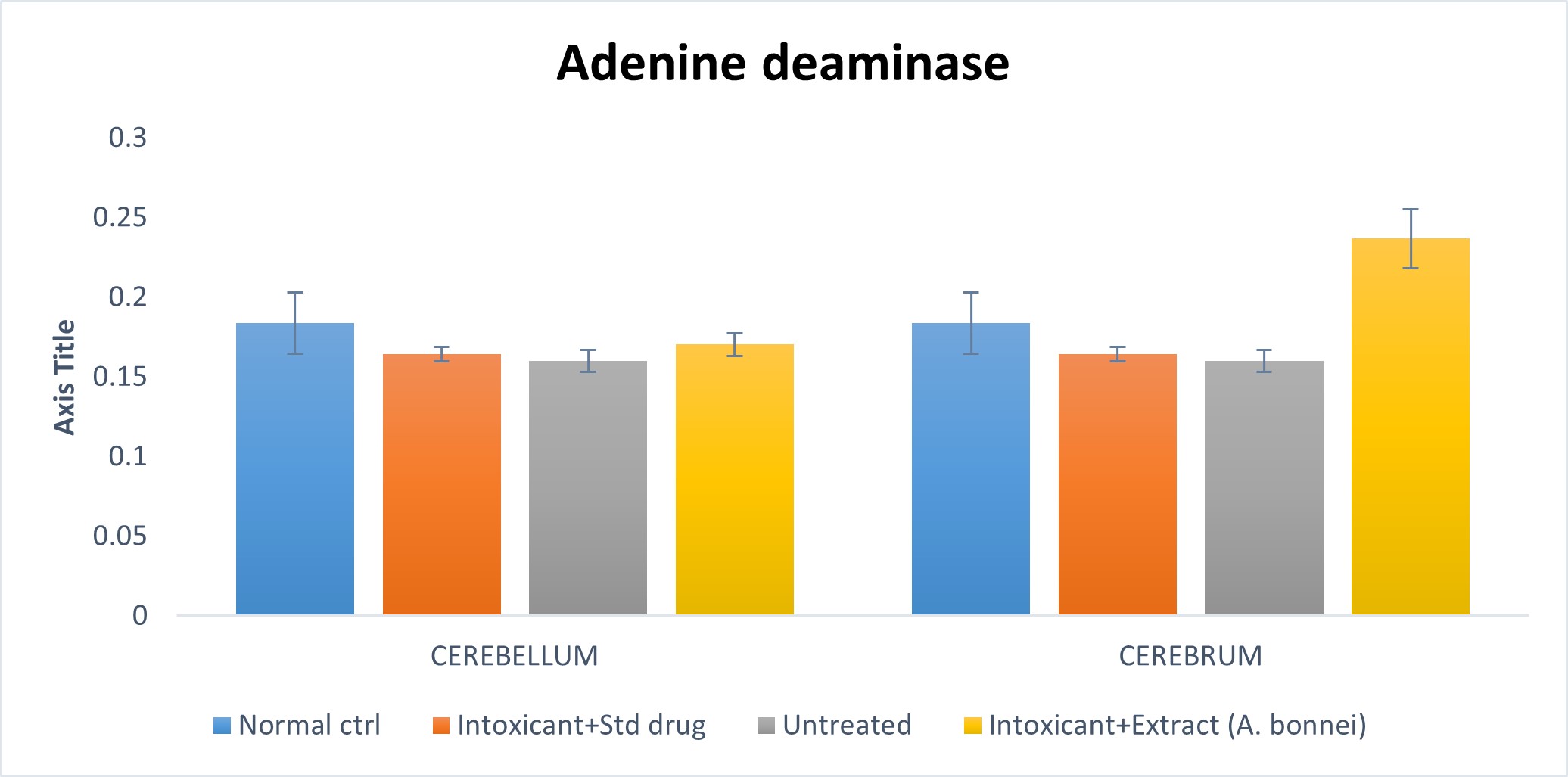
Fig. 4.3: Effect of plant extract of A. bonnei on Adenine deminsase in the cerebellum and cerebrum of Wistar albino rats.
Cerebellum: The chart above shows that an insignificant (p>0.05) decrease existed when the untreated group was compared with the normal control; also the intoxicant + extract group showed an insignificant (p>0.05) decrease on comparism to the normal control. Furthermore, an insignificant (p>0.05) decrease was shown when the untreated group was compared to the intoxicant + standard drug group; while an insignificant (p<0.05) increase was recorded when the intoxicant + extract group was compared to the intoxicant + standard drug.
Cerebrum: Using the chart above, a significant (p>0.05) decrease was shown when the untreated group was compared with the normal control; while a significant (p<0.05) increase was recorded when the intoxicant + extract group was compared to the normal control. Also, an insignificant (p>0.05) decrease was recorded when the untreated was compared to the intoxicant + standard drug group; while a significant increase (p<0.05) was shown for intoxicant + extract group against the intoxicant + standard drug group.
DISCUSSION, CONCLUSION AND RECOMMENDATION
Discussion
Among the compounds of mercury, the methylmercury is primarily responsible for the neurological alterations present in humans and experimental animals. It is believed that the mechanisms are related to the toxic increase in reactive oxygen species (ROS). Oxidative stress is associated with the etiology of neurodegenerative diseases such as amyotrophic lateral sclerosis, Parkinson’s disease, and Alzheimer’s disease (Roulet et al., 1998; Bridges et al., 2010), but these mechanisms have yet to be fully recognized.
From this research, it was seen that the concentration of vitamin E in the cerebrum and cerebellum of the animals were depleted in the untreated group while the group treated with the extract of A. bonnie showed a significant increase when compared with the untreated group. It has been recognized for more than fifty years that adequate levels of vitamin E are critical for maintaining neurological health. The most striking evidence for this requirement is the pathological manifestations of the familial syndrome ataxia with vitamin E deficiency (AVED) in affected humans (Ouahchi et al., 1995, Cavalier et al., 1998). These individuals present with low or non-detectable levels of plasma α-tocopherol and progressive spinocerebellar ataxia, sometimes accompanied by retinitis pigmentosis, proprioception and areflexia (Harding, 1987, Goss-Sampson et al., 1988, Sokol, 1990, Cavalier et al., 1998).
Importantly, pathologic progression can be halted in affected individuals by high-dose supplementation with α-tocopherol (Harding et al., 1985, Matsuya et al., 1994, Amiel et al., 1995, Yokota et al., 1996, Cavalier et al., 1998). Similar findings were established in genetic and dietary models of vitamin E deficiency in mice (Yokota et al., 2001), rats (Einarson, 1953, Goss-Sampson et al., 1988), monkeys (Dinning and Day, 1957) and horses (Mayhew et al., 1987, Aleman, 2011). Vitamin E deficiency-induced neurodegeneration is characterized by axonopathy, demyelination, spheroid formation and defective retrograde transport, prominently affecting the sensory neurons and more specifically the dorsal root ganglion cells (Goss-Sampson et al., 1988, Southam et al., 1991).
Also in this research work, acetylcholinesterase (AChE) was seen to be markedly increased in the intoxicated and untreated group when compared with other groups. This increase was significantly reduced by the plant’s extract as indicated in fig. 4.2. AChE represents one of the first validated biomarkers in environmental and occupational medicine and its use is increased in the last two decades. However, recent findings indicate new potentialities of AChE in human biomonitoring. The sensitivity of AChE activity to other classes of chemicals, including emerging pollutants such as nanomaterials, suggests the usefulness of this biomarker for providing an integrative measurement of the overall neurotoxic risk arising from the whole burden of bioavailable contaminants in areas contaminated by several classes of pollutants.
Environmental pollutants, heavy metals such as mercury, organophosphorus and carbamate pesticides are known to be specific inhibitors of aceylcholinesterase catalytic activity (Ojewole, 1984). They have become the most widely used pesticides today since the removal of organochlorine pesticides from use. AChE belongs to the family of cholinesterases (ChEs), which are specialized carboxylic ester hydrolases that break down esters of choline. Cholinesterase class includes AChE which hydrolyzes the neurotransmitter acetylcholine and pseudocholinesterase or butyrylcholinesterase (BChE)which utilizes butyrylcholine as substrate. AChE is mainly found at neuromuscular junctions and cholinergic synapses in the central nervous system.Here, AChE hydrolyzes acetylcholine into choline and acetate after activation of acetylcholine receptors at the postsynapticmembrane.AChE activity serves to terminate synaptic transmission, preventing continuous nerve firings at nerve endings. Therefore, it is essential for the normal functioning of the central and peripheral nervous system. AChE is also found on the red blood cell membranes, where it constitutes the Yt blood group antigen (Zacharia et al., 2010) also known as Cartwright. It helps to determine a person’s blood type, but the physiological function on erythrocyte membrane is to date unknown (Zacharia et al., 2010).
Furthermore, the results in fig. 4.3 shows that the activity of adenine deaminase in the intoxicated group in both cerebrum and cerebellum of the animals had no significant alteration when compared with the normal control while the extract-treated group showed an increase in its activity when compared with other groups.
Adenosine deaminase plays an important role in controlling adenosine levels. Adenosine modulates the proliferation, survival and apoptosis of many different cell types (Hebert et al., 1999; Retter et al., 2013). Changes in the structure and functionality in the developing brain exposed to methylmercury display a range of effects varying from severe cerebral palsy to subtle developmental delays (Le Roux, 2011; Robertson et al., 2014). In fact, studies have shown that there was a difference in enzyme activity both in distinct areas of the brain and at different stages of development. Adenosine deaminase activity was higher in hippocampus than in cerebral cortex in all groups studied.
Conclusion
In this work, it has been seen that mercury intoxication affects the cerebrum and cerebellum of animals. From these results, it has been observed that the methanol leaves extract of A. bonnei, possesses some neuroprotective properties.
Recommendation
Owing to the ability of the extract to cause significant effects on the parameters assayed herein, it is recommended that
- Further investigations are needed in other higher primate so as to validate the traditional use of the plant in the neurodegenerative disorders.
- Further phytochemical characterization of the plant extract should be done in order to identify the specific compound(s) involved in the observed neuroprotective activities.
REFERENCE
- Abbiw, D. (1990): Useful Plants of Ghana: West African Uses of Wild and Cultivated Plants, Intermediate Technology Publications, Royal Botanical Garden, Kew, London, 1990
- Aebi H. Catalase in vitro: methods in enzymology, 2014; 105: 121-126.
- Agarwal, A., Kanekar, S., Sabat, S., Thamburaj,K.(2016). Metronidazole-induced cerebellar toxicity.Neurology International; 8(6365): 4-6.
- Ahmad, A., M. Chaudhary, A. Soni, A. Payasi and V.K. Dwivedi, 2010. Comparative toxicity profile study of mebatic vs. ofloxacin, ornidazole and metronidazole drugs in rat model. Asian Journal of Biochemistry, pp: 1-11.
- Ahmed and H. Khater, M. (2001). The protective effect of garlic oil on hepatotoxicity induced by acetaminophen in mice and comparison with N-acetylcysteine. Saudi. Med. J. 22: 10801084.
- Akamatsu H, Horio T (2008) The possible role of reactive oxygen species generated by neutrophils in mediating acne inflammation. Dermatology 196: 82–85.
- Al-Dabagh and J. and Mohammad, W. M. (2008). AASLD position paper: the management of acute liver failure. Hepatology 41: 1179-1197.
- Alsheikh-Ali AA, Kuvin JT, Karas RH (2004) Risk of adverse events with fibrates. American Journal of Cardiology 94: 935-938.
- Amacher DE (2002) A toxicologist’s guide to biomarkers of hepatic response. Human Experimental Toxicology 21: 253-262.
- Amar PJ, Schiff ER (2007) Acetaminophen safety and hepatotoxicity – Where do we go from here? Expert Opin Drug Saf 6: 341-355.
- Arora N, Goldhaber SZ (2006) Anticoagulants and transaminase elevation. Circulation 113: 698702.
- Asiedu-Gyekye, B. M, Avaria, M., Basu, P. K. and Wells, P. G. (2014). Pharmacological studies on the in vivo cataractogenicity of acetaminophen in mice and rabbits. Fundamental Applied Toxicology 10: 596-606.
- Barak AJ, Beckenhauer HC, Mailliard ME, Kharbanda KK, Tuma DJ (2003) Betaine lowers elevated S-adenosylhomocysteine levels in hepatocytes from ethanol-fed rats. Journal of Nutrition 133: 2845-2848.
- Bass, N.M., Ockner, B.A., 1996. Drug-induced liver disease. In: Zakin, D, Boyer, T.D. (Eds.), Hepatology, a Text Book of Liver Diseases, 3rd edition W.B. Saunders, Philadephia, pp. 962–1017.
- Belardelli F, Ferrantini M (2002) Cytokines as a link between innate and adaptive antitumor immunity. Trends Immunology 23: 201–208.
- Bergan, T., 2015. Antibacterial activity and pharmacokinetics of nitroimidazole. A review.Scandinavian Journal of Infectious Diseases, 46: 64-71.
- Bhardwaj SS, Chalasani N (2007) Lipid lowering agents that cause druginduced hepatotoxicity. Clin Liver Dis 11: 597.
- Bjornsson, E., Nordlinder, H., Olsson, R. (2002). Metronidazole as a probable cause of severe liver injury.Hepatogastroenterology; 49: 252-254.
- Bleibel W, Kim S, D’Silva K, Lemmer ER (2007) Drug-induced liver injury: Review Article. Dig Dis Sci 52: 2463-2471.
- Burtis, J. T. and Ashwood, G. (2001). Acetaminophen kinetics in acutely poisoned patients. Clinical Pharmacology Ther. 25: 184-195.
- Carlson, I. I. and Mohammad, F. K. (2003).Pharmacokinetics and Distribution of
- Caylor, K.B. and M.K. Cassimatis, (2001). Metronidazole neurotoxicosis in two cats. Journal of the American Animal Hospital Association, 37: 258-262.
- Chandak,S., AgaRwal,A., Shukla,A., Joon,P.(2016). A Case Report of Metronidazole
- Chang C. Y, Schaino TD (2007) Review article: Drug hepatotoxicity. Aliment Pharmacol Ther 25: 1135-1151
- Cheong, M. G., Beyer, J., Hall, G. L., deGraffenried, L. A. and Adams, P. E. (2011). Predictive value of liver slices for metabolism and toxicity in vivo: use of acetaminophen as a model hepatotoxicant. Toxicol. Appl. Pharmacol. 122: 108-116.
- Chitturi S, George J (2002) Hepatotoxicity of commonly used drugs: nonsteroidal antiinflammatory drugs, antihipertensives, antidiabetic agents, psycotropic drugs. Semin Liver Dis 22: 195-206.
- Chukwu V.O., Akudike, C.J., Ezejindu, D.N.,Augustine I.C. (2015) a. The Protective Effects ofTurmeric on Liver Enzymes of Metronidazole TreatedAdult Male Wistar Rats. International Journal of Advancesin Scientific Research; 1(6): 255-258.
- Chukwu V.O., Akudike, C.J., Ezejindu, D.N.,Ofoego, U.C., Chukwuocha, C.C. (2015) b. The ProtectiveEffects of Turmeric on testicular tissues, after treatmentwith metronidazole in adult male Wistar Rats. Journal of Pharmaceutical Sciences; 4 (1) 62-68.
- Chumark, P., Khunawat, P., Sanvarinda, Y.,Phornchirasilp, S., Morales, N.P., et al. (2008). The in vitroand ex vivo antioxidant properties, hypolipidaemic andantiatherosclerotic
- Dambach DM, Andrews BA, Moulin F. (2005). New technologies and screening strategies for hepatotoxicity: use of in vitro models. Toxicology Pathology 33: 17-26.
- Deng X, Luyendyk JP, Ganey PE, Roth R. A. (2009). Inflammatory stress and idiosyncratic hepatotoxicity: Hints from animal models. Pharmacology Revision 61: 262282.
- Evans, J., (2002). The use of diazepam in the treatment of metronidazole toxicosis in the dog. activities of water extract of Moringaoleifera Lam leaves. Journal of Ethnopharmacology 116: 439–446.
- Finch, R.G. and I.S. Snyder, (2016). Antiprotozoan drugs. In: Modern pharmacology. Eds.: C.R. Craige nd R.E. Stitzel. Little, Brown Co.Boston, pp: 729-740.
- Finegold, S.M., (2000). Metronidazole. Annals of Internal Medicine, 93: 585-587.
- Frey, H.H. and W. Löscher, (2016). Lehrbuch der Pharmakologie und Toxikologie für die Veterinar-medizin. Enke, Stuttgart. pp: 502-503.
- Gaur, J. H. and Bhatia, D. E. (2009). Hepatic injury due to drugs, chemicals and toxins. In “MacSween’s Pathology of the Liver”. 5th ed. pp. 649-765. Burt, A. D., Portmann, B. C., and Ferrell, L. D. ed. Churchill Livingstone. New York, U. S. A.
- Giffen, M. J., Alvarez, M., Culebras, J. M. and GonzálezGallego, J. (2002). An overview of animal models for investigating the pathogenesis and therapeutic strategies in acute hepatic failure. World Journal of Gastroenterology 15: 3086-3098.
- Giusti G. and Galanti B. (1984): Colorimetric Method. Adenosine deaminase In: Bergmeyer HU, (ed). Methods of enzymatic Analysis. 3rd ed. Weinheim: Verlag chemie; 1984.
- Godfrey,M. S., Finn, A., Zainah,H., Dapaah-Afriyie,K. (2015): Metronidazole-induced encephalopathyafter prolonged metronidazole course for treatment of C.difficile colitis. B.M.J. Case Reports; doi:10.1136/bcr-2014-206162.
- Groman, R., 2000. Metronidazole. Compend Cont Educ., 22: 1104-1107, 1130.
- Groneberg DA, Grosse-Siestrup C, Fischer A (2002) In vitro models to study hepatotoxicity. Toxicol Pathol 30: 394-399.
- Gulteridge JMC, Wilkin C. Copper dependent hydroxyl radical damage to ascorbic acid formation of a thiobarbituric acid reactive products FEBs Lett, 2000; 137:327-340.
- Gupta, A.K., M.P. Agarwal, R. Avasthi, D.P. Bhadoria and N. Rohatgi, 2003.
- Ikemoto M, Tsunekawa S, Toda Y, Totani M (2001) Liver-type arginase is a highly sensitive marker for hepatocellular damage in rats. Clinical Chemistry 47: 946948.
- InducedNeurotoxicity in Liver Abscess Patient and the Usefulnessof MRI for its Diagnosis. Journal of Clinical and Diagnostic Research. Vol-10(1): TD06-TD07.
- Ju C, Uetrecht JP (2002) Mechanism of idiosyncratic drug reactions: Reactive metabolite formation, protein binding and the regulation of the immune system. Curr Drug Metab 3: 367-377.
- Kafrouni MI, Anders RA, Verma S (2007) Hepatotoxicity associated with dietary supplements containing anabolic steroids. Clinical Gastroenterology Hepatology 5: 809-812.
- Kanbac G, Dokumacioglu A, Tektas A, Kartkaya K, Erden Inal M (2009) Betaine (trimethylglycine) as a nutritional agent prevents oxidative stress after chronic ethanol consumption in pancreatic tissue of rats. International Journal of Vitamin Nutrition Resourse 79: 79-86.
- Kaplowit, M. (2001). Pentoxifylline and N-acetylcysteine in hepatic ischemia/reperfusion injury. Clinical Chimistry Acta 275: 127-135.
- Kapoor, K., Chandra, M., Nag, D., Paliwal, J.K.,Gupta, R.C., Saxena, R.C. (1999). Evaluation ofmetronidazole toxicity: a prospective study. Internation Journal of Clinical Pharmacology. Resource.; 19(3):83-88.
- Khalili H, Dashti-Khavidaki S, Rasoolinejad M, Rezaie L, Etminana M (2009) Anti-tuberculosis drugs related hepatotoxicity: Incidence, risk factors, pattern of changes in liver enzymes and outcome. DARU 17: 163-167.
- Kharbanda KK (2009) Alcoholic liver disease and methionine metabolism. Semin Liver Dis 29: 155165.
- Kozer E, Koren G (2001) Management of paracetamol overdose: Current controversies. Drug Safety 24: 503-512.
- Kumar, H., Sharma, A., Attri,S.K., Kaushik, S.(2012). Rapid onset peripheral neuropathy: A rarecomplication of metronidazole. Journal, Indian Academy ofClinical Medicine; 13(4): 346-348.
- Kumari, M., Singh, P. (2013). Study on thereproductive organs and fertility of the male mice followingadministration of metronidazole. Int. J. Fertil. Steril. 7(3):225-238.
- Langman G, Hall PM, Todd G (2001) Role of non-alcoholic steatohepatitis in methotrexateinduced liver injury. Journal of Gastroenterology Hepatology 16: 1395-1401.
- Lau L., Dodd, S., Dean, O., Copolov, D. L., Malhi, G. S. and Berk, M. (1992). N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert. Opin. Biol. Ther. 8: 19551962.
- Lee J, Boyer JL (2000) Molecular alterations in hepatocyte transport. Seminars in Liver Disease 20: 373-384.
- Ligha, A.E. and C.W. Paul, 2011. Oxidative Effect of metronidazole on The Testes of Wistar Rats.
- Lim AYL, Segarra I, Chakravarthi S, Akram S, Judson JP (2010) Histopathology and biochemistry analysis of the interaction between sunitinib and paracetamol in mice. BMC Pharmacology 10: 14-30.
- Luyendyk JP, Shaw PJ, Green CD, Maddox JF, Ganey PE, et al. (2005) Coagulation-mediated hypoxia and neutrophil-dependent hepatic injury in rats given lipopolysaccharide and ranitidine. Journal of Pharmacology Experimental Therapuetics 314: 1023-1031.
- Lynch T, Price A (2007) The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician 76: 391-396.
- Manov I, Motanis H, Frumin I, Iancu TC (2006) Hepatotoxicity of antiinflammatory and analgesic drugs: Ultrastructural aspects. Acta Pharmacology Sin 27: 259-272.
- Masuda Y (2006) Learning toxicology from carbon tetrachloride-induced hepatotoxicity. Yakugaku Zasshi 126: 885-899.
- MetronidazoleAdministered Intraperitoneally in MicePharmacologyonline 3: 858-863.
- Metronidazole-induced neurotoxicity. Journal of the Association of Physicians of India, 51: 617-618.
- Mochizuki M, Shimizu S, Urasoko Y, Umeshita K, Kamata T, (2009) Carbon tetrachloride-induced hepatotoxicity in pregnant and lactating rats. Journal of Toxicology Science 34: 175-181.
- Mudry, M.D., M.A. Carballo, M.D. Labal de Vinuesa, M. Gonz´alez Cid and I. Larripa, 2014. Mutagenic bioassay of certain pharmacological drugs: III. Metronidazole (MTZ). Mutation Research, 86: 243-77.
- Mukinda, J.T, Eagles, F.K., (2010). Acute and sub-chronic oral toxicity profile of the aqueous extract of Pohygala fruticosa in female mice and rats. Journal of Ethnopharmacology 128, 236–240.
- Murayama N, Reimers A, Hari Y, Hunziker T, Gerber H, Muller U, Pich-ler W (2006) Drug-induced linear IgA bullous dermatosis associated with ceftriaxone- and metronidazole-specific T cells. Dermatology 199: 25–30.
- Naruse, Z., Tang C. and Makuuchi, (2007). Species variation in toxication and detoxication of acetaminophen in vivo: a comparative study of biliary and urinary excretion of acetaminophen metabolites. Journal of Pharmacology and Experimental Therapuetics. 244: 91-99.
- Navarro VJ, Senior JR (2006) Drug-related hepatotoxicity. N Engl J Med 354: 731-739.
- Nishimura Y, Kurata N, Sakurai E, Yasuhara H (2004) Inhibitory effect of antituberculosis drugs on human cytochrome P450-mediated activities. Journal of Pharmacology Science 96: 293300.
- O’Brien PJ, Slaughter MR, Polley SR, Kramer K (2002) Advantages of glutamate dehydrogenase as a blood biomarker of acute hepatic injury in rats. Lab Anim 36: 313-321.
- O’Connor N, Dargan PI, Jones AL (2003) Hepatocellular damage from nonsteroidal antiinflammatory drugs. QJM 96: 787–791.
- Oda, S.S. (2012). Histopathological andbiochemical alterations of metronidazole-induced toxicityin male rats. Global Veterin.,9: 303-310.
- Olson H, Betton G, Robinson D, Thomas K, Monro A, et al. (2000) Concordance of the toxicology of pharmaceuticals in humans and animals. Regulatory Toxicology Pharmacology 32: 5667.
- Oyedeji, K.O., Oshatimi Abayomi, Abidoye Dele,Adeleke K.O. (2015). Effect of Metronidazole onReproductive Parameters in Male Wistar Rats. International Journal of Pharmacological Science Revision Resource., 35(1), 186-190.
- Ozer, G., Aykaç, G., Uysal, M. and Oz, H. (2008). Liver lipid peroxidation and glutathione-related defence enzyme systems in mice treated with paracetamol. Journal of Applied Toxicology 14: 297-299.
- Papay I., Hammond, C. L., Lee, T. K. and Ballatori, N. (2006). Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. J. Hepatol. 34: 946954.
- Park BK, Naisbitt DJ, Gordon SF, Kitteringham NR, Pirmohamed M (2001) Metabolic activation in drug allergies. Toxicology 158: 11-23.
- Parkinson A, Klaassen CD (2001) Biotransformation of xenobiotics. In: Casarett and Doull’s Toxicology. (6th edn) McGraw-Hill, New York.
- Plumb, D.C. (2005). Plumb’s Veterinary DrugHandbook, 5th ed., Ames, Blackwell Publishing.
- Protective effect of diallyl sulfone against acetaminophen-induced hepatotoxicity in mice. Biochem. Toxicol. 11: 11-20.
- Rahman C., and Hodgson,, D. V. (2000). Species variation in the metabolic activation of paracetamol to toxic intermediates: role of cytochromes p-450 and p-448. Toxicol. Lett. 16: 55-61.
- Ramaiah SK (2007) A toxicologist guide to the diagnostic interpretation of hepatic biochemical parameters. Food Chemical Toxicology 45: 1551-1557.
- Ramaiah, S.K., 2011. Preclinical safety assessment. Current gaps, challenges and approaches in identifying translatable biomarkers of drug- induced liver damage. Clin. Lab. Med. 31, 161–172.
- Roach, S.S. (2007). Introductory clinicalpharmacology, 8th ed. Philadelphia: Lippincott, Williams and Wilkins.
- Rogulja, R. R., Hughes, R. D., Bansal, S., Lehec, S. C., Wendon, J. A. and Dhawan, A. (2013). Effects of serum from patients with acute liver failure due to paracetamol overdose on human hepatocytes in vitro. Transplant. Proc. 37: 2391-2394.
- Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- Sahu AJ, Nicholls RJ, Forbes A, Ellis HJ, Ciclitira PJ (2008) Human lymphocyte stimulation with pouchitis flora is greater than with flora from a healthy pouch but is suppressed by metronidazole. Gut 53: 1801–1805.
- Saukkonen K., Jemnitz, K., Veres, Z., Monostory, K., Kóbori, L. and Vereczkey, L. (2006). Interspecies differences in acetaminophen sensitivity of human, rat, and mouse primary hepatocytes. Toxicol. In Vitro 22: 961-967.
- Scorza, A. V. & M. R. Lappin (2004). Metronidazote for the treatment of fetine giardiasis. Journal of Feline Medicine and Surgery 6(3): 157-60.
- Sekis, I., et al. (2009). Single-dose pharmacokinetics and genotoxicity of metronidazole in cats. Journal of Feline Medicine and Surgery 11(2): 60-8.
- Shinn DLS. Metronidazole in acute ulcerative gingivitis. Lancet1962;279:1191.
- Sohrabi, D., M. Alipour and A. Mellati, 2007. Effect of metronidazole on spermatogenesis, plasma gonadotrophins and testosterone in rats. Iranian Journal of Reproductive Medicine, 5: 69-72.
- Stolk MF, Becx MC, Kuypers KC, Seldenrijk CA (2006) Severe hepatic side effects of ezetimibe. Clin Gastroenterol Hepatol 4: 908-911.
- Sulkowski M, Mehta S, Chaisson R, Thomas D, Moore R (2004) Hepatotoxicity associated with protease inhibitor-based antiretroviral regimens with or without concurrent ritonavir. AIDS 18: 2277-2284.
- Tabak, F., R. Ozaras, Y. Erzin, A.F. Celik, G. Ozbay and H. Senturk, 2003. Ornidazole-induced liver damage: Report of three cases and review of the literature. Liver International, 23: 351418.
- Tally FP, Sutter VL, Finegold SM. Metronidazole versus anaerobes: invitro data and initial clinical observations. Calif Med 1972;117:22–6.
- Thapa M. J. and W, I. A. (2001). Acetaminophen overdose. 662 cases with evaluation of oral acetylcysteine treatment. Arch. Intern. Med. 141: 380-385.
- Tong MH, Landers DV, Meyn L, Hillier SL (2005) Clinical and cervical cytokine response to treatment with oral or vaginal metronidazole for bacterial vaginosis during pregnancy: a randomized trial. Obstet Gynecol 102: 527–534.
- Uetrecht J (2006) Role of animal models in the study of drug-induced hypersensitivity reactions. AAPS J 7: 914-921.
- Uetrecht J (2006) Role of animal models in the study of drug-induced hypersensitivity reactions. AAPS J 7: 914-921.
- Vernau, K. (2009). Cerebellar Disease. Veterinary Neurology Symposium; Univ. of Calif.-Davis. accessed via Veterinary Information Network; vin.com
- Wallace J. L (2004) Acetaminophen hepatotoxicity: NO to the rescue. Br J Pharmacol 143: 1-2.
- Willett K. L, Roth R. A, Walker L (2004) Workshop overview: hepatotoxicity assessment for botanical dietary supplements. Toxicol Sci 79: 4-9.
- Williams D. P, Park BK (2003) Idiosyncratic toxicity: The role of toxicophores and bioactivation. Drug Discov Today 8: 1044-1050.
- Wright T. M, Vandenberg AM (2007) Risperidone- and quetiapine-induced cholestasis. Ann Pharmacother 41: 1518-1523.
- Wright, K.H., Tyler J.W. (2005): Recognizingmetronidazole toxicosis in dogs, Vet. Med., 98:410418.
- Yamamoto, M. C., Wang, E. J., Patten, C., Lee, M. J., Xiao, F., Reuhl, K. R. and Yang, C. S. (2012).
- Yu AS, Keeffe EB (2002) Nonalcoholic fatty liver disease. Rev Gastroenterol Disord 2: 11-19.
- Zollner G, Marschall HU, Wagner M, Trauner M (2006) Role of nuclear receptors in the adaptive response to bile acids and cholestasis: pathogenetic and therapeutic considerations. Mol Pharm 3: 231-251.
- Zollner G, Marschall HU, Wagner M, Trauner M (2006) Role of nuclear receptors in the adaptive response to bile acids and cholestasis: pathogenetic and therapeutic considerations. Mol Pharm 3: 231-251.