
Pattern of Hemoglobin F in Children with Sickle Cell Disease Attending a Tertiary Hospital in Southwest Nigeria.
- Nosimot O. Davies
- Leona U. Njoku
- Ademola S. Adewoyin
- 886-894
- Jun 15, 2024
- Health
Pattern of Hemoglobin F in Children with Sickle Cell Disease Attending a Tertiary Hospital in Southwest Nigeria.
*1Nosimot O. Davies, 2Leona U. Njoku, 1Ademola S. Adewoyin.
1Department of Haematology and blood Transfusion, College of Medicine, University of Lagos.
2Department of Haematology, University of Lagos.
*Corresponding Author
DOI: https://doi.org/10.51244/IJRSI.2024.1105056
Received: 25 April 2024; Revised: 13 May 2024; Accepted: 17 May 2024; Published: 15 June 2024
ABSTRACT
Background: Sickle cell disease (SCD) is the most common form of an inherited haemoglobinopathy, accounting for about 70% of the world’s major haemoglobinopathies, and the greatest burden is in sub-Saharan Africa. Fetal haemoglobin (HbF) delays the polymerization of Sickle cell disease. In Nigeria, there is no recent data on the levels of haemoglobin F and its association with SCD.
Aim: This study evaluated the levels of HbF of 76 children with SCD attending Lagos University Teaching Hospital (LUTH) and correlated the levels with their age.
Methods: Using a BIO-RAD D10 hgh-performance liquid chromatography machine, HbF levels of seventy-six (76) SCD children 70 HbSS and 6 HbSC was determined. The relationship between HbF levels and SCD was assessed using the SPSS version 22.0 (Statistical Package for Social Sciences) for statistical analysis.
Result: The mean HbF of the 76 participants with SCD was (12.9 ± 5.57). The mean HBF level was significantly higher in children aged 0-5 years (14.83 ± 4.65) was significantly higher than older age group (6-10 years = 6.78 ± 1.93; 11-15 years = 4.74 ± 1.14; > 15 years = 4.37 ± 1.01), p = 0.000. There was a negative correlation between HBF and age of participants (r = -0.7942)
Conclusion: There is an inverse relationship between HbF level and the age of children with SCD. HBF levels are highest in children between 0-5 years compared to older age groups. This finding will assist clinicians especially in the need for HBF-inducing agents such as hydroxyurea in managing these children.
Keywords: Sickle Cell Disease, Haemoglobin F, Haemoglobin SS, Haemoglobin SC,
INTRODUCTION
Sickle cell disease (SCD) is the most common form of an inherited haemoglobinopathy, accounting for about 70% of the world’s major haemoglobinopathies. (1)
It is characterized as an autosomal, recessive, heterogeneous, and a monogenetic disorder caused by an A-to-T point mutation in the β-globin gene responsible for the production of abnormal hemoglobin S (HbS), which polymerizes in the deoxygenated state and results in the sickling of erythrocytes. Haemoglobin variants are mutant forms of haemoglobin in a population usually occurring as a result they could affect the structure, behavior, the production rate and the stability of the specific gene. Well-known haemoglobin variants such as sick-cell anaemia are responsible for diseases and are considered haemoglobinopathies. (2)
According to the WHO report, hemoglobin disorders are originally endemic to 61% of the 229 countries around the world and 5.2% of the world population carries a significant hemoglobin variant. Again approximately 1.1% of couples are at risk for having children with a hemoglobin disorder and 2.7 per 1000 conceptions are affected worldwide. (3) Approximately 70% of these cases occur in sub-Saharan Africa. (4)
The greatest burden of SCD is in sub-Saharan Africa, with over 300,000 babies being born with SCD annually; this number is expected to increase to up to 400,000 individuals by 2050. (5) Nearly 90 percent of the world’s SCD populations live in three countries: Nigeria, India, and the Democratic Republic of Congo where the disease affects up to 2 percent of the population, and the carrier prevalence rate is as high as 10 to 30%. (6)
Nigeria alone has been estimated to have at least 150,000 newborns born with SCD annually. Estimates are challenging because of the lack of federal newborn screening programs; however, approximately 700,000 births occur per year and the prevalence of SCD in newborns was 3% in a regional newborn screening program. (7)
Due to the high prevalence of sickle cell trait at 23.7%, the frequency of sickle cell anaemia in Nigeria is about 20 per 1000 births resulting in about 150,000 babies being born each year with the disorder. (4) This makes Nigeria the country with the largest burden of sickle cell disease globally with 4.1% of the population affected. (8)
Haemoglobin is responsible for the transport of oxygen from the lung to the tissue and carries carbon dioxide from the tissue to the lung for gaseous exchange. It also serves as a buffer to maintain the pH of the physiology milieu of the body. (9) There are different types of haemoglobin and their globin chain composition varies at different stages of life. During fetal life, Gower I, Portland, Gower II, and fetal Hb predominate in uterine life there is a transition to adult Hb within 3-6 months of life. (10)
The phenotypic heterogeneity of sickle cell disease is strongly modulated by foetal haemoglobin. (11) Reduced rate of acute painful episodes, leg ulcers, osteonecrosis, acute chest syndromes, and reduced disease severity have all been associated with elevated levels of Hb F while the association of complications like stroke and priapism with Hb F is unclear. (12)
These could point to the fact that Hb F levels are influenced by β-globin haplotypes and genetic polymorphisms. For instance, Senegal and Arab-Indian haplotypes usually have the highest Hb F and mildest clinical presentations while Bantu haplotypes have the lowest Hb F and most severe clinical course; Benin haplotypes have intermediate levels of Hb F and moderate clinical course. (13)
Varied foetal haemoglobin levels (2-9%) have been reported from different studies involving sickle cell patients in Nigeria. (14-18)
Due to paucity of data, the pattern of hemoglobin F in sickle cell disease amongst children in Lagos Nigeria is unknown. The purpose of this research is to provide recent, valuable, and accurate clinical information on the pattern of fetal hemoglobin in children with sickle cell disease and to correlate the levels of haemoglobin F with sickle cell disease.
METHODS
Study Site
The study was conducted at the Sickle Cell Paediatrics Clinic of the Lagos University Teaching Hospital (LUTH) over six months. Lagos University Teaching Hospital is a tertiary hospital located in Idi-Araba, Surulere Lagos State Nigeria, and is affiliated with the College of Medicine of the University of Lagos.
Study Design
The study was a cross-sectional hospital-based study.
Sample Size Determination
The sample size was determined using the formular:
N= (Zα + Zβ)2 Pο (1- Po) + P1 (1-P1)2 / (P1-Po)2
N = Minimum sample size in each group
Po= Response in the first group from previous study = 0.98 (19)
P1= expected response in the second group = 0.80
Zα = % of normal distribution corresponding to the required significant level of 5% = 1.96 (95% confidence interval).
Zβ = point of normal distribution corresponding to the statistical power of 80%= 0.842
N = (1.96 +0.842) 2 0.98(1-0.98) + 0.80 (1- 0.8)2 ÷ (0.80-0.98)2
7.85 X 0.0196 + 0.032 / 0.00324
N = 57. 36 (approximately 57)
However, to accommodate unforeseen errors and attrition and also increase the power of the study, a total number of 76 patients were recruited for the study.
Participant Selection
A total of 76 participants were recruited. This includes patients with haemoglobinopathy between the ages of 2 to 16 attending the pediatric sickle cell clinic of LUTH Idi Araba whose parents consented to participate in the study by signing the consent form after adequate information was given to them.
Inclusion Criteria
Inclusion criteria included SCD patients aged 2-16 years attending the pediatric sickle cell clinic of LUTH.
Exclusion Criteria
Exclusion criteria included Known SCD children whose parents have declined consent to partake in the study. Patients on disease-modifying drugs such as hydroxyurea, patients with beta thalassemias and hereditary persistence of fetal hemoglobin which causes hemoglobin F levels to be higher than normal and non SCD children between 2 years and above 16 years were excluded.
Sample Collection and Processing.
A consecutive, non- purposive sampling technique was used.
Under aseptic conditions, 2 mls of blood sample was collected from the median cubital vein by venipuncture and dispensed into an EDTA sample bottle it was carefully mixed.
The fetal hemoglobin levels assay was carried out using an automated BIO-RAD DIO high- performance liquid chromatography (HPLC) machine. These analyses were done within 3 hours of sample collection.
Fetal Haemoglobin Assay: Elution Method (High-Performance Liquid Chromatography)
Principle: When whole blood is passed between a mobile phase (eluent) and a stationary phase, the specific intermolecular reactions between the molecules of whole blood and the packing material of the column cause different constituents of the sample to elute at different times which brings about separation. This is measured by a UV detector which recognizes converts and records the analytes after they leave the column. This is then interpreted through computer software and shown in a chromatogram. The result is read in percentages and values noted down in a booklet.
Statistical Analysis
The data obtained was entered into the computer and analyzed using the statistical software package for Social Sciences (SPSS), version 22. Descriptive statistics (frequencies, means and standard deviation) were generated. Comparison between continuous variables was done using the student’s t-test or ANOVA as appropriate.
The correlation was determined using Pearson’s correlation coefficient. A p-value less than 0.05 was statistically significant.
Ethical Clearance and Informed Consent
Ethical approval was obtained from the Lagos State University Teaching Hospital Research Ethics Committee before the research commenced. Written consent was obtained from each parent, or caregiver after a brief and vivid explanation of the research details. Assent was then obtained from the participating children or wards. The parents or caregiver reserved the right to withdraw their child or ward from the research whenever they desired without consequences.
RESULTS
Table 1 shows the socio-demographic characteristics of the study participants.
A total of 76 children were recruited. This comprised 43 (56.58%) males and 33 (43.42%) females. Also, most of the participants 70 (92.11%) were HbSS patients (39 males, 31 females) and 6 (7.89%) were HbSC patients (4 males, 2 females).
Table 1. Socio-demographic characteristics of respondents
| Variable | Subject (n=76) | Percentage (%) |
| Age group
0-5 years 6-10 years 11-15 years >15 years |
29
18 23 6 |
38.16
23.68 30.26 7.89 |
| Education
Nursery Primary Secondary |
29
18 29 |
38.16
23.68 38.16 |
| Religion
Christian Muslim Others |
20
49 7 |
26.3
64.47 9.2 |
| Mean age of participant
Male Female |
43.2 ± 7.86
39.85 ± 0.15 |
|
| Sex
Male Female |
43
33 |
56.60
43.40 |
| Genotype
SS SC |
70
6 |
92.0
7.80 |
| Mean age of study participants (6.54±5.06)
Mean age of SC participants (5.83± 3.97) Mean age of SS participants (8.52± 5.14) |
||
Table 2 shows the comparison of the haemoglobin variants by age range of the participants. The total mean Fetal hemoglobin for all the sickle cell disease patients is 12.9± 5.57. The mean haemoglobin F decreased as the age increased, and this was statistically significant.
| Age range of participant | N | Mean ± Std. Deviation | P-Value | |
| HbA | 0-5 | 29 | 2.91 ± 1.09 | |
| 6-10 | 18 | 3.63 ± 1.11 | 0.006 | |
| 11-15 | 23 | 3.91 ± 0.90 | ||
| >15 | 6 | 3.63 ± 0.93 | ||
| HbS | 0-5 | 29 | 75.85 ± 14.32 | |
| 6-10 | 18 | 81.61 ± 13.86 | 0.02 | |
| 11-15 | 23 | 86.15 ± 6.05 | ||
| >15 | 6 | 87.30 ± 2.83 | ||
| HbA2 | 0-5 | 29 | 1.95 ± 0.99 | |
| 6-10 | 18 | 2.49 ± 1.19 | 0.000 | |
| 11-15 | 23 | 3.30 ± 0.89 | ||
| >15 | 6 | 3.70 ± 1.23 | ||
| HbF | 0-5 | 29 | 14.83 ± 4.65 | |
| 6-10 | 18 | 6.78 ± 1.93 | 0.000 | |
| 11-15 | 23 | 4.74 ± 1.14 | ||
| >15 | 6 | 4.37 ± 1.013 | ||
Table 2: Comparing levels of Haemoglobin variants per age group across the participants
Table 3 show the mean and standard deviation of Haemoglobin variants in the sickle cell disease patients. The mean level of HBF is higher in HBSC patients compared to HBSS patients. The diffence was however not statistically significant (p = 0.537).
Table 3: Mean and standard deviation of Haemoglobin variants in Sickle Cell Disease
| Haematological parameters | HbSS | HbSC | P-value |
| HbF | 8.93 ± 5.250 | 10.40 ± 9.079 | 0.537 |
| HbA2 | 2.74 ± 1.160 | 1.26 ± 0.722 | 0.003* |
| HbS | 84.48 ± 4.244 | 43.33 ± 13.246 | 0.001* |
| HbA | 3.560 ± 1.046 | 2.047 ± 0.632 | 0.001* |
Note: * = Significant difference
Figure 1 shows the Pearson’s linear correlation graph between HBF and the age of the participants. There is a negative correlation between HbF and age. The Pearson linear correlation coefficient is (Y= 0.5846) and the linear regression coefficient (r= -0.7942)
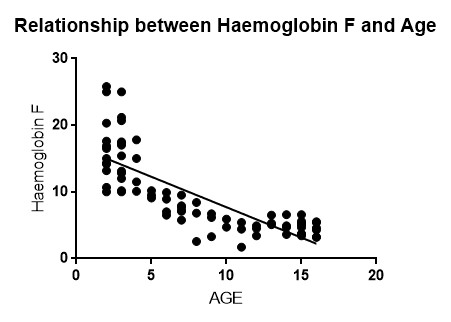
Pearson’s correlation coefficient, (Y= 0.5846) and the linear regression coefficient (r= -0.7942)
Fig 1: scatter plot of Haemoglobin F versus age of participants.
DISCUSSION
The exclusion of fetal hemoglobin (HbF) from the sickle hemoglobin polymer is the major genetic modulator of the hematologic and clinical features of sickle cell disease. Clinical observations, epidemiologic studies, biophysical measurements, and therapeutic trials have all confirmed these beneficial effects. (20) In this study, 76 participants who were patients attending the pediatric sickle cell clinic of the Lagos University Teaching Hospital, with mean age of 6.54 ± 5.06 years were examined to determine the Pattern of HBF in Sickle Cell Disease. This study is similar to the study of Akinlosotu et al. on the relationship between fetal haemoglobin and haematological indices in children with sickle cell anaemia from Southwestern Nigeria where mean age observed was 7.6 ± 3.5. (21) However, this finding contradicts the report by Mpalampa et al. on fetal haemoglobin and diseases severity in sickle cell anaemia patients in Kampala, Uganda, where mean of age observed was 9.3 ± 4.8 years. (22)
The mean level of HBF of 12.91 ± 5.57 observed in this study, is much higher that the mean values reported from previous studies from Nigeria. Adeolu et al, reported a mean HBF level of 9.9 ± 6.0% among 105 children with SCD in Ile-Ife, southwestern Nigeria (23), while a mean of 2.99 ± 5.16 was reported from Sokoto, North-west Nigeria. (24) Furthermore, the mean HbF level in this study is much higher than the value of 7.2 ±5.0 reported by Tshilolo et al in Congo. (11) These disparities could be attributed to a difference in the method of estimation foetal hemoglobin, difference in study population and possibly geographical variation. A much lower value of 5.16±4.04 was also reported from a previous study involving adult sickle cell patients in Ibadan south-western Nigeria. (25) Most participants with high HbF levels (38.16%) were between the ages of 0-5 years which is consistent with findings from a study conducted in Kampala, Uganda among sickle cell anaemia patients in 2012. (26) This study showed that children who are 5 years and below had significantly higher level of HBF compared to older age group. The lowest mean value was in children above 15 years old. This finding corroborates the finding from various studies that HBF levels decrease with increasing age. This decrease in HbF level with age was observed in a study where fetal hemoglobin (HbF) waned at 6 months after birth but persisted in some adults. (27)
This study also demonstrated a negative correlation between the level of HbF and age. This indicates an inverse relationship age and HBF levels and is similar to reports from earlier studies indicating that HbF usually disappears gradually from infants’ red blood cells after about 6 months. (28) The findings of this study indicate that fetal hemoglobin levels are significantly higher in children 0-5 years, moderated in children 6-10 years and lower in children 11-16 years. Lower fetal haemoglobin levels in these children were also significantly related to advancing age.
This study is limited by its relatively small sample size and being limited to a single centre. The findings may therefore not be generalizable. A larger scale, possibly multi-centre study is desired.
CONCLUSION
Fetal haemoglobin can alter the pathophysiology and clinical course and provide prospective curative therapy to sickle cell disease- a primary disorder of red blood cells- inherited in an autosomal recessive fashion.
The findings of this research show an inverse relationship between HbF and the age of children with sickle cell disease attending the Lagos University Teaching Hospital. y, HbF levels are high in children 0-5 years, moderate with children aged 6-10 years and low in children aged 11-15 years. This finding will assist clinicians especially in the need for HBF-inducing agents such as hydroxyurea in managing these children.
Competing Interests
There are no competing interests.
REFERENCES
- Hoffbrand AV, Moss PAH, Moss Petit JE. Genetic disorders of haemoglobin: In Essential Haematology, 5th edition. (2006). UK, Blackwell Publishing Ltd. 6:72- 93.
- Barabino GA, Platt MO, Kaul DK. (2010). Sickle Cell biomechanics. Annual Reviews of Biomedical Engineering. 12: 345–367)
- Modell B, Darlison M. (2008) Global epidemiology of hemoglobin disorders and derived service indicators. Bull World Health Organ. 86:417– 496
- World Health Organisation. (2006). Sickle cell disease in the African Region: Current situation and the way forward. WHO Africa Regional Report. AFR/RC/56/17.
- Poludasu S, Ramkissoon K, Salciccioli L, Kamran H, Lazar JM. (2013) Left ventricular systolic function in sickle cell anemia: a meta-analysis. Journal of Cardiac Failure. 19:333–341
- Sachdev V, Machado RF, Shizukuda Y, Rao YN, Sidenko S, et al. (2007). Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. Journal of American College Cardiology. 49:472–479
- Lundberg JO, Gladwin MT, Weitzberg E. (2015). Strategies to increase nitric oxide signalling in cardiovascular disease. Nature Reviews Drug Discovery. 14:623–641.
- Omotade OO, Kayode CM, Falade SL, Ikpeme S, et al. (1998). Routine screening for sickle cell haemoglobinopathy by electrophoresis in an infant welfare clinic. West African Journal of Medicine.17(2):91-94.
- Lichtman MA, Kipps TJ, Seligsohn U, Kaushansky K, Paschal TJ. (2010) Disorders of haemoglobin structure: Sickle cell anaemia & related abnormalities in Williams Haematology, 8th edition. Landsteiner publications Chapter.48:871-914.
- Hoffbrand AV, Moss PAH, Moss Petit JE. (2006) Genetic disorders of haemoglobin: In Essential Haematology, 5th edition, UK, Blackwell Publishing Ltd. 6:72- 93.
- Tshilolo L,Summa V, Gregorj C,Kinsiama C, Bazeboso JA, Avvisati G, Labie D. (2012). Foetal Haemoglobin, Erythrocytes Containing Foetal Haemoglobin, and Hematological Features in Congolese Patients with Sickle Cell Anaemia. Hindawi Publishing Corporation: Anaemia. 105349:1-7.
- Akinsheye I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastian P, Chui DHK, Steinberg MH. (2011) Foetal haemoglobin in sickle cell anaemia. Blood. 118(1):19-27.
- Steinberg MH. (2009). Genetic etiologies for phenotypic diversity in sickle cell anaemia. Scientific World Journal. 9: 46-67.
- Akanni EO, Oseni BS, Bamisaye EO, Raji AA, Mewoyeka OO, Hassan RO. (2011). Haemoglobin F level in different haemoglobin variants. Korean Journal of Hematology. 46(2):118-122.
- Isah IZ, Udomah FP, Erhabor O, Aghedo F, Uko EK, Okwesili AN, et al. (2013). Foetal haemoglobin levels in sickle cell disease patients in Sokoto, Nigeria. British Journal of Medical and Health Sciences. 1(6):36-47.
- Enosolease ME, Ejele OA, Awodu OA. (2005). The influence of foetal haemoglobin on the frequency of vaso-occlusive crisis in sickle cell anaemia patients. Niger Postgrd Med J. 12(2): 102- 105.
- Fatunde OJ, Scott-Emuakpor AB. (1993). Haemoglobin F and A2 in Nigerian children with sickle cell anaemia. J Trop Pediatr. 1993; 39(4): 251-252.
- Kotila TR, Fawole OI, Shokumbi WA. (2000) Haemoglobin F and clinical severity of sickle cell anaemia among Nigerian adults. African Journal of Medicine and Medical Science. 29(3- 4):229-231.
- Nwenyi E, Leafman J, Mathieson K, Ezeobah N. (2014). Differences in quality of life between pediatric sickle cell patients who used hydroxyurea and those who did not. International Journal of Health Care Quality Assurance. 27(6):468–481.
- WHO/AFRO. (2014). The health of the people: what works – the African Regional Health Report. Public health – organization and administration.
- Akinlosotu MA, Adegoke SA, Oseni SB, Adeodu OO. (2017). Relationship between foetal haemoglobin and haematological indices in children with sickle cell anaemia from Southwestern Nigeria. Niger Postgraduate Medical Journal. 24:195-200.
- Mpalampa L, Ndugwa CM, Ddungu H, Idro R. (2012). Foetal haemoglobin and disease severity in sickle cell anaemia patients in Kampala, Uganda. BMC Blood Disorders. 12:11.
- Adeolu OO, Akinsolotu MA, Adegoke SA, Oseni SBA. (2017). Foetal Haemoglobin and Disease Severity in Nigerian Children with Sickle Cell Anaemia. Mediterr J Hematol Infec Dis. 9 (1): e2017063.
- Isah IZ, Udomah FP, Erhabor O, Aghedo F, Uko EK, Okwesili AN, et al. (2013) Foetal haemoglobin levels in sickle cell disease patients in Sokoto, Nigeria. British Journal of Medical Health Science. 1:36‑47.
- Olaniyi JA, Arinola OG, Odetunde AB. (2010) Foetal Haemoglobin (HbF) status in adult sickle cell anaemia patients in Ibadan Nigeria. Annals of Ibadan Postgraduate medical Journal. 8(1): 30-33.
- Mpalampa L, Ndugwa CM, Ddungu H, Idro R. (2012). Foetal haemoglobin and disease severity in sickle cell anaemia patients in Kampala, Uganda. BMC Blood Disorder. 12:11.
- Dominic E, Charles AB, Domnic A. (2006). Fetal haemoglobin during infancy and in sickle cell adults. African health sciences. 6(1): 51-54.
- Martyres DJ, Vijenthira A, Barrowman N, Harris-Janz S, Chretien C, Klaassen RJ. (2009) Nutrient insufficiencies/deficiencies in children with sickle cell disease and its association with increased disease severity. Pediatric Blood. 5(6)720-725